46
Содержание:
Триплоидии
Крайне редко наблюдаемые при мертворождениях, триплоидии составляют пятую по частоте хромосомную аномалию в материале выкидыше. В зависимости от соотношения половых хромосом может быть 3 варианта триплоидий: 69XYY (самая редкая), 69, XXX и 69, XXY (самая частая). Анализ полового хроматина показывает, что при конфигурации 69, XXX чаще всего обнаруживается только одна глыбка хроматина, а при конфигурации 69, XXY чаще всего половой хроматин не обнаруживается.
Приведенный ниже рисунок иллюстрирует различные механизмы, приводящие к развитию триплоидии (диандрию, дигинию, диспермию). С помощью специальных методов (хромосомные маркеры, антигены тканевой совместимости) удалось установить относительную роль каждого из этих механизмов в развитии триплоидии у зародыша. Оказалось, что на 50 случаев наблюдений триплоидия была следствием дигинии в 11 случаях (22%), диандрии либо диспермии — в 20 случаях (40%), диспермии — в 18 случаях (36%).
| Механизмы образования триплоидной зиготы |
|---|
Описание
Молекулярно-цитогенетическая диагностика распространённых хромосомных нарушений (анеуплоидий) по 13, 16, 18, 21, 22, X, Y-хромосоме — молекулярно‐цитогенетический метод, позволяющий определять изменения хромосом анеуплоидий (число хромосом, не кратное гаплоидному набору).
Половые хромосомы — X и Y, и аутосомы — 13, 16, 18, 21, 22 — наиболее часты и имеют наиболее негативное влияние на пренатальное развитие человека. Молекулярно-цитогенетическая диагностика
Молекулярно-цитогенетическая диагностика позволяет выявить отклонение в количестве хромосом (анеуплоидия). В эмбрионах с анеуплоидией отсутствует одна хромосома (моносомия), или имеется лишняя хромосома (трисомия). Наиболее распространённой хромосомной патологией является трисомия (три хромосомы вместо двух в норме) по 21-й хромосоме, или синдром Дауна. Возможно рождение живых детей с трисомиями по хромосоме 13 (синдром Патау), хромосоме 18 (синдром Эдвардса). Эти хромосомные болезни характеризуются еще более тяжёлыми, чем при синдроме Дауна, пороками умственного и физического развития
Как правило, такие дети не живут более года.Спонтанное прерывание беременности
Нарушения репродуктивной функции женского организма, которая сопровождается неожиданным прерыванием беременности, могут быть обусловлены действием различных факторов, среди которых важное место занимают генетические. Во многих случаях беременность прерывается из-за хромосомных нарушений при образовании эмбриона, несовместимых с его жизнью
Чаще возникают ситуации, когда в результате нарушения овогенеза (созревания яйцеклетки) или сперматогенеза (формирования зрелых сперматозоидов) возникают клетки с утраченной или лишней хромосомой или образуется зигота (зародыш) с набором хромосом, по количеству превышающим норму (в норме зигота содержит 46 хромосом). Причинами этому могут быть хронический стресс, курение, употребление наркотиков, алкоголя, экологически неблагоприятные ситуации, а также внутренние факторы: наличие в организме матери или отца генов — небольших участков хромосом, предрасполагающих к нерасхождению хромосом в процессе формирования половых клеток. А когда формируется генетически неполноценный зародыш, по законам естественного отбора организм матери пытается избавиться от него.
С помощью молекулярно-цитогенетической диагностики распространённых хромосомных нарушений, исследуя хорион, определяют отклонения в хромосомном наборе. Данные исследования могут быть выполнены, в том числе и новорождённым детям, в этом случае исследуется кровь плода.
На более позднем сроке прерывания беременности проводится исследование наиболее часто встречающихся анеуплодий (изменений числа хромосом) при неразвивающейся беременности по 13, 16, 18, 21, 22, X, Y-хромосомам (7 хромосом).Показания:
- На более поздних сроках прерывания беременности.
- Беременные от 35 лет и старше.
- Наличие в семье ребёнка с хромосомными заболеваниями или врождёнными пороками развития.
- Замершая беременность с установленными хромосомными дефектами у плода.
- В семье есть ребёнок с хромосомной патологией; носителем хромосомной патологии является один из членов семьи.
- У ближайших родственников есть моногенные заболевания (например, муковисцидоз).
- Женщины, перед зачатием или на раннем сроке беременности принимали сильнодействующие препараты (например, противоопухолевые) или перенесли такие инфекции, как гепатит, краснуха, токсоплазмоз.
- Членам семей с отягощённым акушерским анамнезом (невынашивание, самопроизвольные аборты, мертворождение, рождение ребёнка с врождёнными пороками).
- Высокий риск рождения ребенка с хромосомной патологией, рассчитанный по результатам комбинированного скрининга (УЗИ+биохимические тесты).
- Пороки или отклонения развития, выявленные при ультразвуковом исследовании (ультразвуковые маркёры хромосомной патологии).
Подготовка
Условия подготовки к анализу определяются лечащим врачом.
Синдром Ангельмана
Патофизиология
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала по 21-й хромосоме, либо полностью (трисомия), либо частично (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от степени копии, генетической истории и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например был обнаружен у обезьян и мышей). Совсем недавно[когда?
] исследователи[кто? ] вывели трансгенных мышей с наличием 21-й человеческой хромосомы (в дополнение к стандартному набору мышей). Добавление генетического материала может проводиться в разных направлениях. Типичный человеческий кариотип обозначается как 46,XY (мужской) или 46,XX (женский) (различие в поле несёт Y-хромосома).
Трисомия
Трисомия — это наличие трёх гомологичных хромосом вместо пары в норме.
Синдром Дауна и сходные хромосомные аномалии чаще встречаются у детей, рождённых немолодыми женщинами. Точная причина этого неизвестна, но, по-видимому, она как-то связана с возрастом яйцеклеток матери.
Трисомия происходит из-за того, что во время мейоза хромосомы не расходятся. При слиянии с гаметой противоположного пола у эмбриона образуется 47 хромосом, а не 46, как без трисомии.
Трисомия 21-й хромосомы в 95 % случаев является причиной возникновения синдрома Дауна, и в 88 % случаев из-за нерасхождения материнских гамет и в 8 % — мужских.
Мозаицизм
Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, XX/47, XX, 21). Данная форма синдрома является как правило более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики.
По данному типу синдром появляется в 1—2 % случаев.
Робертсоновские транслокации
Дополнительный материал 21-й хромосомы, вызывающий синдром Дауна, может появиться за счёт робертсоновских транслокаций в кариотипе одного из родителей. В данном случае длинное плечо 21-й хромосомы прикреплено к плечу другой хромосомы (чаще всего 14-й ). Фенотип у человека с робертсоновскими транслокациями соответствует норме. Во время репродукции, нормальный мейоз повышает шанс на трисомию 21-й хромосомы и рождения ребёнка с синдромом Дауна. Транслокации с синдромом Дауна часто называют семейный синдром Дауна
. Это не зависит от возраста матери и показывает скорее равную роль родительских организмов в появлении синдрома Дауна. Данный тип появления синдрома занимает 2—3 % от всех случаев.
Дублирование части хромосомы 21
Очень редко, но части хромосом могут делиться. Это создаст дополнительные копии некоторых, но не всех генов из 21-й хромосомы. Если продублируются фрагменты, обусловливающие физические и психологические проявления синдрома Дауна, то ребёнок родится с этим синдромом. Такое дублирование происходит крайне редко и не существует оценки периодичности данного явления.
Формы синдрома Дауна
Примерно в 91 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка.
Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет.
Что гены могут рассказать о вашем здоровье
С помощью генетического тестирования можно узнать о рисках для здоровья еще до того, как они превратились в проблему. Но тест выявляет не только наследственные заболевания. Если вы планируете ребенка, в первую очередь стоит задуматься о здоровье в целом, ведь оно еще понадобится, чтобы растить детей.
Генетический тест Атлас анализирует образцы ДНК и ответы в опроснике, чтобы составить полный профиль здоровья. На основе этих результатов вы получите рекомендации, которые помогут улучшить показатели здоровья.
| Раздел | |
|---|---|
| Риски заболеваний | Оценка риска развития 20 многофакторных заболеваний |
| Статус наследственных заболеваний | Оценка риска статуса носительства 318 наследственных заболеваний |
| Питание и метаболизм | Влияние генетики на способность организма синтезировать витамины, минералы, нутриенты; а также вкусовые предпочтения, метаболизм кофеина, непереносимость лактозы и глютена |
| Спорт и риски спортивных травм | Влияние ДНК на ваш метаболизм при занятиях спортом и риски получения спортивных травм |
| Рекомендации | Индивидуальные рекомендации, направленные на снижение рисков заболеваний и улучшение показателей здоровья |
Узнать больше о своих генах, чтобы учесть это при планирвоании семьи можно, заказав Генетический тест Атлас на нашем сайте.
- World Health Organisation, Genes and human diseases
- Nemours KidsHealth, The Basics on Genes and Genetic Disorders
- ScienceDaily, Human Genome
- Cystic Fibrosis Foundation, About Cystic Fibrosis
- Seyed Bashir Mirtajani et al, Geographical distribution of cystic fibrosis; The past 70 years of data analysis, 2017
Заболевания и расстройства
Следующие заболевания относятся к числу заболеваний, связанных с генами на хромосоме 12:
- ахондрогенез 2 типа
- коллагенопатия II и XI типов
- плоская роговица 2
- эпизодическая атаксия
- наследственная геморрагическая телеангиэктазия
- гипохондрогенез
- буллезный ихтиоз Сименса
- Самая известная дисплазия
- Синдром Кабуки
- зрелый диабет у молодых людей 3-го типа
- метилмалоновая ацидемия
- нарколепсия
- несиндромная глухота
- Синдром Нунана
- болезнь Паркинсона
- Синдром Паллистера-Киллиана ( тетрасомия 12р)
- фенилкетонурия
- спондилоэпиметафизарная дисплазия по типу Штрудвика
- врожденная спондилоэпифизарная дисплазия
- спондилопериферическая дисплазия
- Синдром Стиклер , ( COL2A1 о связанных)
- Заикание
- Дефицит триозофосфат-изомеразы
- тирозинемия
- Болезнь фон Виллебранда
Зачем знать о рисках наследственных заболеваний
Человеческий организм состоит из триллионов клеток. Каждая играет определенную роль: отвечает за производство ферментов для расщепления пищи, транспортировку кислорода в крови или выполняет другие необходимые для жизни функции.
Большинство клеток человека содержит 23 хромосомы от матери и 23 — от отца. Всего 46 хромосом
Гены — участки ДНК, которые содержатся в хромосомах и передаются по наследству. У человека по разным оценкам 20–25 тысяч генов. Они изменяются при передаче потомству — во время деления клетки получают по одной хромосоме от каждой пары.
Изменения в генах, которые приводят к нарушению работы важных белков и развитию заболеваний, называются патогенными вариантами генов. В зависимости от степени изменений в генах заболевания делят на следующие категории:
| Категория | Причина развития | Примеры заболеваний |
|---|---|---|
| Хромосомные заболевания | Вся хромосома или ее большие сегменты отсутствуют, дублируются или изменены. | Синдром Дауна, трисомия 13, трисомия 18, синдром Клайнфельтера и синдром Тернера |
| Моногенные заболевания | В гене происходит изменение, из-за которого он перестает работать | Муковисцидоз, серповидноклеточная анемия, синдром ломкой Х-хромосомы, мышечная дистрофия или болезнь Хантингтона |
| Многофакторные расстройства | Мутации нескольких генов. Часто в сочетании с факторами окружающей среды | Диабет, болезнь Альцгеймера, Паркинсона, ожирение |
| Митохондриальные нарушения | Редкие нарушения, вызванные вариантами гена в митохондриальной ДНК. Эти нарушения могут поражать любую часть тела, в том числе мозг и мышцы. | Болезнь Альцгеймера, мышечная дистрофия, болезнь Лу Герига, диабет и рак |
Наличие патогенного варианта гена только у матери или отца, не означает, что у ребенка обязательно проявится заболевание. Человек может быть здоровым носителем. Но если у обоих родителей одинаковый патогенный вариант гена, то риск рождения ребенка с заболеванием значительно повышается.
Одно из самых распространенных наследственных заболеваний в европейской популяции — муковисцидоз, при котором в легких и других органах скапливается слизь. Она блокирует легкие, поджелудочную железу и другие органы. По статистике в Европе один из 2 000–3 000 новорожденных появляется на свет с муковисцидозом. Только половина пациентов с этим диагнозом доживает до 18 лет, и только 5% — до 40 лет.
Наследственные заболевания часто не совместимы с жизнью, поэтому распространено мнение о том, что они не так часто встречаются. Но по статистике 1 из 50 людей рождается с моногенным заболеванием, в то время как у 1 из 263 есть хромосомные нарушения.
В мире живет более 70 000 людей с муковисцидозом.
При наличии у обоих родителей варианта гена, связанного с муковисцидозом, риск рождения ребенка с этим заболеванием — 25%. Если узнать об этом заранее с помощью генетического теста, можно вовремя принять необходимые меры, например, воспользоваться вспомогательными репродуктивными технологиями.
Анатомические данные
Анализ материала самопроизвольных выкидышей, сбор которого был начат в начале двадцатого века в Институте Карнеги, позволил выявить огромный процент аномалий развития среди абортусов ранних сроков
В 1943 году Хертиг и Шелдон опубликовали результаты патологоанатомического исследования материала 1000 выкидышей на ранних сроках. Материнские причины невынашивания беременности были ими исключены в 617 случаев. Современные данные указывают на то, что мацерированные зародыши во внешне нормальных оболочках тоже могут быть связаны с хромосомными аномалиями, что в сумме составляет около 3/4 всех случаев данного исследования.
| Морфологическое исследование 1000 абортусов (по Hertig и Sheldon, 1943) | ||
| Грубые патологические нарушения плодного яйца:
плодное яйцо без зародыша или с недифференцированным зародышем |
489 | |
| Локальные аномалии зародышей | 32 | |
| Аномалии плаценты | 96 | 617 |
| Плодное яйцо без грубых аномалий | ||
| с мацерированными зародышами | 146 | |
| 763 | ||
| с немацерированными зародышами | 74 | |
| Аномалии матки | 64 | |
| Другие нарушения | 99 |
Дальнейшие исследования Микамо и Миллера и Полланда позволили уточнить связь между сроком выкидыша и частотой нарушений развития зародыша. Оказалось, что чем меньше срок выкидыша, тем частота аномалий выше. В материалах выкидышей, происшедших до 5-й недели после зачатия макроскопические морфологические аномалии плодного яйца встречаются в 90% случаев, при сроке выкидыша от 5 до 7 недель после зачатия — в 60%, при сроке больше 7 недель после зачатия — менее, чем в 15—20%.
Важность значения остановки развития зародыша в ранних самопроизвольных выкидышах была показана прежде всего фундаментальными исследованиями Артура Хертига, который в 1959 г. опубликовал результаты исследования человеческих зародышей до 17 дней после зачатия
Это был плод его 25-летней работы.
У 210 женщин в возрасте до 40 лет, идущих на операцию гистерэктомии (удаления матки) дата операции была сопоставлена с датой овуляции (возможного зачатия). После операции матки подвергались самому тщательному гистологическому исследованию на предмет выявления возможной беременности малого срока. Из 210 женщин только 107 были оставлены в исследовании в связи с обнаружением признаков овуляции, и отсутствием грубых нарушений труб и яичников, препятствующих наступлению беременности. Было обнаружено 34 плодных яйца, из них 21 плодное яйцо было внешне нормальным, а 13 (38%) имело явные признаки аномалий, которые, по мнению Хертига, обязательно привели бы к выкидышу или на этапе имплантации или вскоре после имплантации. Поскольку в то время не было возможности проведения генетического исследования плодных яиц, причины нарушений развития зародышей оставались неизвестными.
При обследовании женщин с подтвержденной фертильностью (все пациентки имели по несколько детей) было обнаружено, что одно из трех плодных яиц имеет аномалии и подвергается выкидышу до появления признаков беременности.
Этиология и причины нарушения
Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, бесплодны. Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий за счет нарушения в женских половых клетках, встречается в три раза чаще. Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип. Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизучены. В отличие от других хромосомных заболеваний, влияние возраста родителей отсутствует или выражено незначительно.
Трисомии
В материале выкидышей трисомии представляют более половины всех количественных хромосомных аберраций
Обращает на себя внимание то, что в случаях моносомий недостающей хромосомой обычно оказывается X-хромосома, а в случаях избыточных хромосом, дополнительная хромосома чаще всего оказывается аутосомой
Точная идентификация дополнительной хромосомы стала возможна благодаря методу G-бэндинга. Исследования показали, что все аутосомы могут принимать участие в нон-дисджанкшн (см. таблицу)
Обращает на себя внимание, что три хромосомы, чаще всего встречающиеся при трисомиях новорожденных (15-я, 18-я и 21-я) чаще всего обнаруживаются и при летальных трисомиях у зародышей. Вариации относительных частот различных трисомий у зародышей отражают во многом сроки, на которых происходит гибель зародышей, поскольку, чем более летальной является комбинация хромосом, тем на более ранних сроках происходит остановка развития, тем реже будет обнаруживаться такая аберрация в материалах выкидышей (чем меньше срок остановки развития, тем труднее обнаружить такой зародыш)
| Лишняя хромосома при летальных трисомиях у зародыша (данные 7 исследований: Буэ (Франция), Карр (Канада), Кризи (Великобритания), Дилл (Канада), Кадзи (Швейцария), Такахара (Япония), Теркелсен (Дания)) | ||
|---|---|---|
| Дополнительная аутосома | Количество наблюдений | |
| A | 1 | |
| 2 | 15 | |
| 3 | 5 | |
| B | 4 | 7 |
| 5 | ||
| C | 6 | 1 |
| 7 | 19 | |
| 8 | 17 | |
| 9 | 15 | |
| 10 | 11 | |
| 11 | 1 | |
| 12 | 3 | |
| D | 13 | 15 |
| 14 | 36 | |
| 15 | 35 | |
| E | 16 | 128 |
| 17 | 1 | |
| 18 | 24 | |
| F | 19 | 1 |
| 20 | 5 | |
| G | 21 | 38 |
| 22 | 47 |
Анализ кариотипа
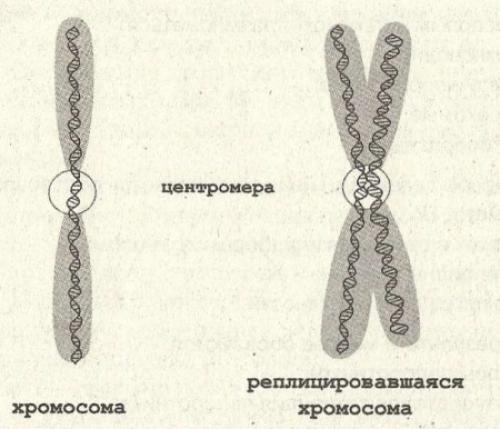
Кариотип – систематизированный набор хромосом ядра клетки с его количественными и качественными характеристиками.
Нормальный женский кариотип — 46,XX Нормальный мужской кариотип — 46,XY
Исследование кариотипа — процедура, призванная выявить отклонения структуры строения и числа хромосом.
Показания для кариотипирования:
- Множественные врожденные пороки развития, сопровождаемые клинически анормальным фенотипом или дизморфизмом
- Умственная отсталость или отставание в развитии
- Нарушение половой дифференцировки или аномалии полового развития
- Первичная или вторичная аменорея
- Аномалии спермограммы – азооспермия или тяжелая олигоспермия
- Бесплодие неясной этиологии
- Привычное невынашивание
- Родители пациента со структурными хромосомными аномалиями
- Повторное рождение детей с хромосомными аномалиями
К сожалению, с помощью исследования кариотипа можно определить лишь крупные структурные перестройки. В большинстве же случаев аномалии строения хромосом представляют собой микроделеции и микродупликации невидимые под микроскопом. Однако такие изменения хорошо идентифицируются современными молекулярными цитогенетическими методами — флуоресцентной гибридизацией (FISH) и хромосомным микроматричным анализом.
Свернуть лишнее
— Кто еще входит в группу риска по возникновению генетических заболеваний?
— Кровнородственные браки. У них выше риск того, что встретятся два «поломанных» гена. У людей, которые не связаны родственными узами, вероятность того, что столкнутся два гена с одной мутацией, равна два случая к 30 тысячам. В группе риска и те, у кого в родне были генетические заболевания.
— В чем сложность лечения генетических болезней? Почему ученые до сих пор не могут найти ключ к восстановлению поломанных участков ДНК?
— Они стараются, работают над тем, чтобы заменить большой кусок ДНК здоровым, но пока есть проблема его доставки в ген без повреждения соседних участков. Если мы говорим о болезни Дауна, казалось бы, надо просто свернуть лишнюю хромосому, чтобы она не работала. Но по последним данным в головном мозге ребенка проходят изменения, похожие на болезнь Альцгеймера, если после рождения попытаться свернуть лишнюю хромосому. В любом случае «выстрелит» в другом месте. Но работа в этом направлении кипит, и я уверена, что скоро мы научимся управлять генами.

Исследования ученых. Фото: pixabay.com
— Но ведь некоторые редкие генетические заболевания уже научились лечить, например, появился укол от спинально-мышечной атрофии?
— Есть орфанные заболевания, когда не хватает определенных ферментов в ДНК. Это несколько другое. Такие уколы содержат эти недостающие ферменты, чтобы попытаться переработать то, что не перерабатывается в организме. Но они не меняют ДНК, они всего лишь замещают недостающее вещество.
Статья по теме
Путевка в жизнь. Как людей с психическими болезнями учат жить в обществе
Пока единственное, чем можно помочь людям с генетическими заболеваниями — поддерживать их состояние. Например, при фенилкетонурии назначается специальное питание. Всю жизнь такие больные едят безбелковую пищу. Мясо, рыбу, яйца без фениламина. Это ограничения навсегда, но по-другому пока нельзя.
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается. Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде. Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания. В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит. В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Психологические особенности
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень. Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким IQ испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции. Такие особенности причиняют пациентам много неприятностей в период обучения и нередко продолжают сказываться на профессиональной деятельности.
Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью. Есть данные, свидетельствующие о склонности пациентов с синдромом Клайнфельтера к гомосексуализму, алкоголизму и наркомании. Сложно сказать, вызваны ли особенности психики у таких больных непосредственным влиянием хромосомной аномалии, или же это реакция на проблемы в сексуальной сфере.
В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных Х-хромосом увеличивается количество и выраженность патологических симптомов.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции. У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью. Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Известные заболевания хромосомной природы
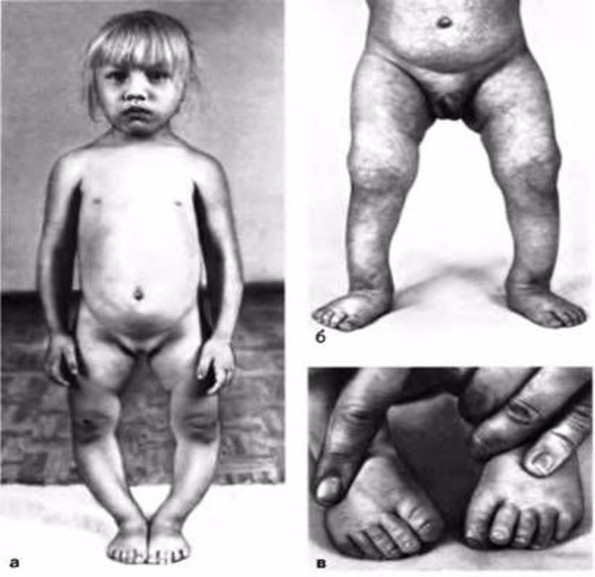
Одним из самых известных заболеваний, происходящих по причине наличия аномалий в генетическом материале, является синдром Дауна. Он обуславливается трисомией по 21 хромосоме. Характерным признаком этой болезни является отставание в развитии. Дети испытывают серьезные проблемы во время обучения в школе, часто им требуется альтернативная методика преподавания материала. Вместе с тем отмечаются нарушения физического развития – плоское лицо, увеличенные глаза, клинодактилия и другие. Если такие люди прикладывают значительные усилия, они могут достаточно хорошо социализироваться, известен даже случай успешного получения высшего образования мужчиной с синдромом Дауна. У больных повышен риск заболеть деменцией. Это и ряд других причин приводит к небольшой продолжительности жизни.
К трисомии относится и синдром Патау, только в этом случае имеется три копии 13 хромосомы. Для заболевания характерны множественные пороки развития, часто с полидактилией. В большинстве случаев отмечается нарушение деятельности центральной нервной системы либо ее неразвитость. Часто (примерно в 80 процентах) больные имеют пороки развития сердца. Тяжелые нарушения приводят к высокой смертности – в первый год жизни умирает до 95% детей с этим диагнозом. Заболевание не поддается лечению или коррекции, как правило, можно лишь обеспечить достаточно постоянный контроль состояния человека.
Еще одна форма трисомии, с которой рождаются дети, относится к 18 хромосоме. Заболевание в этом случае носит название синдрома Эдвардса и характеризуется множественными нарушениями. Деформируются кости, часто наблюдается измененная форма черепа. Сердечно-сосудистая система обычно с пороками развития, также проблемы отмечаются с органами дыхания. В результате около 60% детей не доживают до 3 месяцев, к 1 году умирает до 95% детей с этим диагнозом.
Трисомия по другим хромосомам у новорожденных практически не встречается, поскольку почти всегда приводит к преждевременному прерыванию беременности. В части случаев рождается мертвый ребенок.
С нарушениями числа половых хромосом связан синдром Шерешевского-Тернера. Из-за нарушений в процессе расхождения хромосом теряется X-хромосома в женском организме. В результате организм не получает должного количества гормонов, поэтому нарушается его развитие. В первую очередь это относится к половым органам, которые развиваются лишь отчасти. Практически всегда для женщины это обозначает невозможность иметь детей.
У мужчин полисомия по Y или X хромосоме приводит к развитию синдрома Клайнфельтера. Для этого заболевания характерна слабая выраженность мужских признаков. Зачастую сопровождается гинекомастией, возможно отставание в развитии. В большинстве случаев наблюдаются ранние проблемы с потенцией и бесплодие. В этом случае, как и для синдрома Шерешевского-Тернера, выходом может стать экстракорпоральное оплодотворение.
Цитогенетическая полоса
Идеограммы G-бэндинга 4-й хромосомы человека
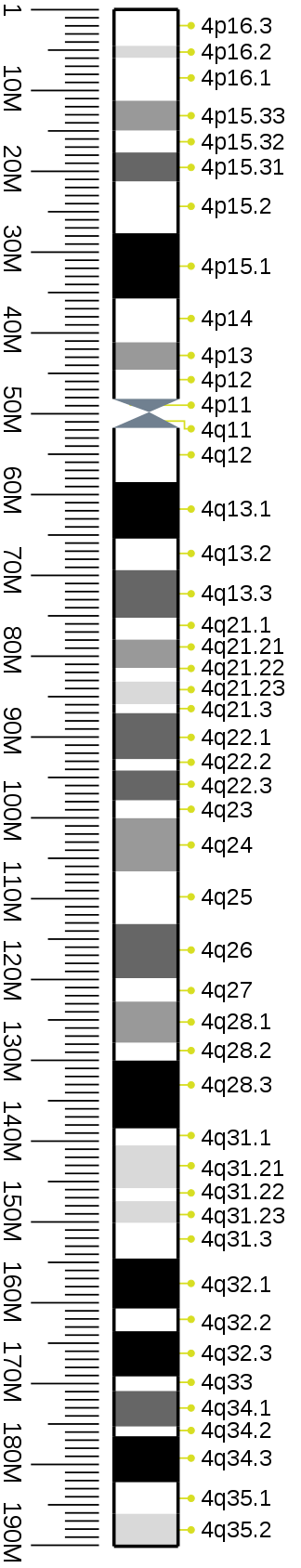
Идеограмма G-бэндинга 4-й хромосомы человека в разрешении 850 ударов в час. Длина полосы на этой диаграмме пропорциональна длине пары оснований. Этот тип идеограммы обычно используется в браузерах генома (например, Ensembl , UCSC Genome Browser ).
Шаблоны G-полос на хромосоме 4 человека в трех различных разрешениях (400, 550 и 850). Длина полосы на этой диаграмме основана на идеограммах из ISCN (2013). Этот тип идеограммы представляет собой действительную относительную длину полосы, наблюдаемую под микроскопом в различные моменты митотического процесса .
| Chr. | Рука | Группа | ISCN начало |
Остановка ISCN |
пар оснований старта |
Остановка базовой пары |
Пятно | Плотность |
|---|---|---|---|---|---|---|---|---|
| 4 | п | 16,3 | 220 | 1 | 4500000 | гнег | ||
| 4 | п | 16,2 | 220 | 389 | 4 500 001 | 6 000 000 | gpos | 25 |
| 4 | п | 16.1 | 389 | 779 | 6 000 001 | 11 300 000 | гнег | |
| 4 | п | 15,33 | 779 | 1066 | 11 300 001 | 15 000 000 | gpos | 50 |
| 4 | п | 15.32 | 1066 | 1286 | 15 000 001 | 17 700 000 | гнег | |
| 4 | п | 15.31 | 1286 | 1557 | 17 700 001 | 21 300 000 | gpos | 75 |
| 4 | п | 15,2 | 1557 | 1811 г. | 21 300 001 | 27 700 000 | гнег | |
| 4 | п | 15.1 | 1811 г. | 2166 | 27 700 001 | 35 800 000 | gpos | 100 |
| 4 | п | 14 | 2166 | 2505 | 35 800 001 | 41 200 000 | гнег | |
| 4 | п | 13 | 2505 | 2742 | 41 200 001 | 44 600 000 | gpos | 50 |
| 4 | п | 12 | 2742 | 2877 | 44 600 001 | 48 200 000 | гнег | |
| 4 | п | 11 | 2877 | 3046 | 48 200 001 | 50 000 000 | Acen | |
| 4 | q | 11 | 3046 | 3249 | 50 000 001 | 51 800 000 | Acen | |
| 4 | q | 12 | 3249 | 3571 | 51 800 001 | 58 500 000 | гнег | |
| 4 | q | 13,1 | 3571 | 3910 | 58 500 001 | 65 500 000 | gpos | 100 |
| 4 | q | 13,2 | 3910 | 4062 | 65 500 001 | 69 400 000 | гнег | |
| 4 | q | 13,3 | 4062 | 4333 | 69 400 001 | 75 300 000 | gpos | 75 |
| 4 | q | 21,1 | 4333 | 4502 | 75 300 001 | 78 000 000 | гнег | |
| 4 | q | 21,21 | 4502 | 4671 | 78 000 001 | 81 500 000 | gpos | 50 |
| 4 | q | 21,22 | 4671 | 4739 | 81 500 001 | 83 200 000 | гнег | |
| 4 | q | 21,23 | 4739 | 4874 | 83 200 001 | 86 000 000 | gpos | 25 |
| 4 | q | 21,3 | 4874 | 5145 | 86 000 001 | 87 100 000 | гнег | |
| 4 | q | 22,1 | 5145 | 5517 | 87 100 001 | 92 800 000 | gpos | 75 |
| 4 | q | 22,2 | 5517 | 5636 | 92 800 001 | 94 200 000 | гнег | |
| 4 | q | 22,3 | 5636 | 5890 | 94 200 001 | 97 900 000 | gpos | 75 |
| 4 | q | 23 | 5890 | 6059 | 97 900 001 | 100 100 000 | гнег | |
| 4 | q | 24 | 6059 | 6347 | 100 100 001 | 106 700 000 | gpos | 50 |
| 4 | q | 25 | 6347 | 6685 | 106 700 001 | 113 200 000 | гнег | |
| 4 | q | 26 год | 6685 | 7040 | 113 200 001 | 119 900 000 | gpos | 75 |
| 4 | q | 27 | 7040 | 7277 | 119 900 001 | 122 800 000 | гнег | |
| 4 | q | 28,1 | 7277 | 7565 | 122 800 001 | 127 900 000 | gpos | 50 |
| 4 | q | 28,2 | 7565 | 7734 | 127 900 001 | 130 100 000 | гнег | |
| 4 | q | 28,3 | 7734 | 8259 | 130 100 001 | 138 500 000 | gpos | 100 |
| 4 | q | 31,1 | 8259 | 8581 | 138 500 001 | 140 600 000 | гнег | |
| 4 | q | 31,21 | 8581 | 8733 | 140 600 001 | 145 900 000 | gpos | 25 |
| 4 | q | 31,22 | 8733 | 8851 | 145 900 001 | 147 500 000 | гнег | |
| 4 | q | 31,23 | 8851 | 9004 | 147 500 001 | 150 200 000 | gpos | 25 |
| 4 | q | 31,3 | 9004 | 9207 | 150 200 001 | 154 600 000 | гнег | |
| 4 | q | 32,1 | 9207 | 9545 | 154 600 001 | 160 800 000 | gpos | 100 |
| 4 | q | 32,2 | 9545 | 9681 | 160 800 001 | 163 600 000 | гнег | |
| 4 | q | 32,3 | 9681 | 9985 | 163 600 001 | 169 200 000 | gpos | 100 |
| 4 | q | 33 | 9985 | 10087 | 169 200 001 | 171 000 000 | гнег | |
| 4 | q | 34,1 | 10087 | 10341 | 171 000 001 | 175 400 000 | gpos | 75 |
| 4 | q | 34,2 | 10341 | 10408 | 175 400 001 | 176 600 000 | гнег | |
| 4 | q | 34,3 | 10408 | 10628 | 176 600 001 | 182 300 000 | gpos | 100 |
| 4 | q | 35,1 | 10628 | 10967 | 182 300 001 | 186 200 000 | гнег | |
| 4 | q | 35,2 | 10967 | 11170 | 186 200 001 | 190 214 555 | gpos | 25 |