Анализ кариотипа (1 чел.)
Содержание:
Причины множественных врожденных пороков развития
Все этиологические факторы МВПР можно разделить на две группы:
- эндогенные — включают в себя генные, геномные и хромосомные мутации, заболевания эндокринной системы, метаболические нарушения, возраст родителей;
- экзогенные — физические (травмы), химические (лекарственные препараты, промышленная и бытовая химия) и биологические (вирусы, бактерии) агенты.
Эти причины приводят к хромосомным, генным и геномным нарушениям, влияют на процессы клеточного и тканевого развития. Большое число множественных врожденных пороков (МВПР) отличаются типом наследования, патогенезом, клиническими проявлениями, прогнозом для пациента.
Лечение синдрома

К сожалению, излечение данного синдрома невозможно.
Терапия, которая может проводиться, обычно направлена на снятия остроты проявляющихся симптомов болезни и предотвращение возникновения сопутствующих заболеваний, составляющих действительно огромный список:
- пневмония, хронический бронхит, астма;
- хронический отит;
- онкологические заболевания разных органов (например, рак горла);
- синусит;
- повышенное или пониженное давление.
Можно отметить, что большинство сопутствующих заболеваний приобретает хронический характер. Чтобы этого не происходило, врачи стараются применять разного рода терапию, если это возможно.
Медикаментозное лечение
Медикаментозное лечение наиболее актуально при синдроме Эдвардса у новорожденных. В терапию включают антибактериальные, противовоспалительные и, чаще всего, гормональные препараты. Они предотвращают развитие некоторых заболеваний, облегчая жизнь ребенку.
Гормональная терапия улучшает функционирование щитовидной железы в организме, что тоже немаловажно для пациента с таким диагнозом
Хирургия
Хирургическое вмешательство при столь серьезной болезни считается нерациональным, так как это может привести к осложнениям или критическому состоянию. Поэтому врачи проводят хирургическое лечение при трисомии 18 только в случае осложнения каких-либо других заболеваний у больного ребенка.
Домашние средства
Учитывая тот факт, что даже медикаментозное лечение данной болезни неэффективно, говорить о лечении дома практически не имеет смысла. Родители могут изготавливать отвары из различных травяных сборов в случае повышенной эмоциональности или агрессивности ребенка. Это поможет ему успокоиться.
Кроме того, часто используются отвары, способные укрепить иммунитет больного ребенка, который ввиду своей болезни, становится очень уязвим к вирусным заболеваниям.
Примеры множественных врожденных пороков развития (МВПР)
- Синдром Дауна. Развивается в результате трисомии по 21-й хромосоме. Пациенты имеют характерные черты лица, страдают задержкой умственного и речевого развития, у них нередко выявляется мышечная гипотония, катаракта, пороки сердца. Средняя продолжительность жизни составляет около 50 лет.
- Синдром Эдвардса. Данная разновидность множественных врожденных пороков развития (МВПР) обусловлена трисомией по 18-й хромосоме. У пациентов выявляется нарушение строения черепа, отсутствие или сужение слухового прохода, широкая грудная клетка, пороки сердечно-сосудистой системы, гипоплазия мозжечка, слабый тонус мускулатуры, судороги, тяжелая умственная отсталость. Характеризуется высокой летальностью.
- Синдром Марфана. Возникает при мутации в гене, который кодирует образование фибриллина. Данное соединение входит в состав практически всех эластичных волокон, поэтому у пациентов отмечается патология опорно-двигательного аппарата, органов зрения, сосудов. При регулярном и правильном лечении продолжительность жизни не снижается.
Другие виды множественных врожденных пороков развития (МВПР) оказывают существенное влияние на жизнедеятельность человека. При этом лечение возможно только симптоматическое, так как изменить дефекты в генах или хромосомах невозможно.
Методы диагностики
Диагностика множественных врожденных пороков развития (МВПР) может осуществляться дородовыми и послеродовыми способами. В первом случае проводятся скрининговые исследования, которые включают в себя УЗИ плода и анализ крови на специфические белки. Такое сочетание позволяет выявить большое количество множественных врожденных пороков развития (МВПР). Врач также может назначать дополнительные методы диагностики: биопсию ворсин хориона и амниоцентез. В этом случае получают не кровь матери, а материал плода, который затем отправляют на генетическое исследование.
Лечение синдрома Эдвардса
Цель терапии: скорректировать опасные для жизни ребенка пороки развития. Но стоит помнить, что у ребенка могут быть серьезные нарушения, и он вряд ли доживет до 12-месячного возраста. Если обнаружена пневмония, то младенцу вводят лекарства от воспаления и антибиотики. Если обнаружено, что у малыша нет сосательного и глотательного рефлекса, то его кормят с помощью зонда. При обнаружении у больного атрезии анала или кишечника нужно восстановить проход пищи.
Если врач видит, что течение синдрома Эдвардса благоприятное, то нужно сделать операцию, чтобы устранить пупочную грыжу, паховую грыжу, сердечные пороки, волчью пасть. Некоторые симптомы можно убрать приемом лекарств. Например, если у младенца запоры, ему понадобятся определенные слабительные лекарства. При скоплении газов в кишечнике назначаются препараты из ряда «пеногасителей».
Дети, у которых выявлена рассматриваемая трисомия, имеют риск таких болезней:
- мочеполовые инфекции
- синуситы и фронтиты
- гипертензия артериальная
- апноэ
- гипертензия легочная
- воспаление легких
- рак почки
- средний отит
Прогноз
Около 95% таких беременностей не заканчиваются рождением живого ребенка. Основные причины смерти включают апноэ и сердечные аномалии. Предсказать точный прогноз при беременности или в периоде новорожденности невозможно . Половина живых младенцев не доживает до первой недели жизни. Средний срок службы составляет от пяти до 15 дней. Около 8–12% младенцев живут дольше 1 года. Один процент детей доживает до 10 лет. Однако эти оценки могут быть пессимистичными; ретроспективное канадское исследование 254 детей с трисомией 18 продемонстрировало 10-летнюю выживаемость 9,8%, а другое обнаружило, что 68,6% детей с хирургическим вмешательством выжили в младенчестве.
Симптомы
Семейной паре нужно понимать, что такое болезнь эдвардса, и какие страдания она способна принести родителям и ребенку. Симптоматика крайне не радостна. Первые признаки будут давать о себе знать уже после рождения:
- Асфиксия.
- Волчья пасть.
- Опущение век (птоз).
- Слабоумие.
- Нарушение глотания.
- Нарушение дыхания.
- Сердечные и сосудистые пороки.
- Заболевания системы пищеварения.
- Нарушения работы мочеполовой системы.
- Проблемы с психикой.
Учитывая все перечисленные симптомы, можно подвести итог, что больной ребенок никогда не станет полноценным членом общества, как бы этого не хотелось. Жизненный путь у него будет коротким, наполненным постоянными преградами. При этом больной не будет понимать и осознавать своего тяжелого состояния из-за тяжелой степени олигофрении.
Но это далеко не все проявления. Эдвардс описал свыше ста тридцати тяжелых симптомов данного недуга.
Скрининг на генетические заболевания
Сегодня известно более 11 000 моногенных заболеваний, которые кодируются одним геном (генетически обусловленные) и передаются от родителей их потомкам. Механизм передачи многих генетических болезней объясняется принципами Менделя.
Аутосомно-доминантные моногенные синдромы встречаются с частотой 1: 200 индивидов; заболевание наблюдается у многих поколений, передается потомкам и рецидивирует с частотой 50%. Примерами аутосомно-доминантных моногенных расстройств могут быть:
- ахондроплазия,
- нейрофиброматоз,
- синдром Марфана,
- болезнь Хантингтона,
- семейный полипоз.
Появление аутосомно-доминантных заболеваний у новорожденных от «здоровых» родителей может быть обусловлено следующими причинами:
1. Мозаицизм зародышевых клеток. Мутация может иметь место лишь в популяции зародышевых клеток. Итак, родители являются непораженными, но могут передавать мутацию потомкам.
2. Новые мутации. Рост возраста родителей ассоциируется с увеличением риска аутосомно-доминантных расстройств (ахондроплазии, танатофорной дисплазии, нейрофиброматоза, синдрома Аперта — краниосиностоз). Риск рецидивов у других детей не увеличивается.
3. Вариабельна экспрессия. Тяжесть заболевания может варьировать, и родители могут не распознать мягкие и субклинические мутации.
4. Уменьшенная пенетрантность. Родители могут иметь аномальный ген без клинических проявлений заболевания.
5. Неверное отцовство. Частота неверного отцовства достигает 15%.
Аутосомно-рецессивные моногенные заболевания проявляются в многочисленных родственников при наличии двух пораженных аллелей. Если оба родителя являются носителями пораженного гена, риск заболевания у потомства равен 25% при каждой беременности. Аутосомно-рецессивные заболевания включают кистозный фиброз, серповидно-клеточную анемию, фенилкетонурию, болезнь Тея-Сакса, Канавана и др.
При Х-сцепленных рецессивных синдромах (гемофилия и др.) мать-носитель пораженного гена передает его своим сыновьям. Итак, 50% сыновей могут быть больными и 50% дочерей будут носителями этого гена. Редкие Х-доминантные синдромы могут передаваться от каждого родителя каждому ребенку подобно аутосомно-доминантных синдромов. Фенотип может сильно варьировать, что связано со смешанной пенетрантностью, лионизацией (гетерохроматизацией) Х-хромосомы (синдром ломкой Х-хромосомы) и геномным импринтингом.
Экспансия тринуклеотидных повторов. Некоторые гены содержат участки тройных повторов (например, ССС). Такие участки являются нестабильными и могут увеличиваться в следующих генерациях, этот феномен получил название антиципации. Количество повторений определяет степень поражения индивида. Экспансия тринуклеотидных повторов составляет основу многочисленных генетических расстройств, таких как синдром ломкой (фрагильной) Х-хромосомы, миотоническая дистрофия и болезнь Хантингтона.
Синдром ломкой (фрагильной) Х-хромосомы является наиболее частой причиной семейной задержки умственного развития. Пораженные мужчины имеют типичные черты: большие уши, выступающая челюсть, большие яички, аутичное поведение, легкая или умеренная умственная отсталость. Женщины обычно менее поражены в связи с инактивацией Х-хромосомы.
Ген ломкой Х-хромосомы локализуется в Х-хромосоме и имеет три нуклеотидные повтора (ССС). Нормальные индивиды имеют 6-50 повторов, непораженные носители женского пола могут иметь 50-200 повторов, которые могут распространяться на мейоза до полной мутации при наличии более 200 повторов. Если имеет место полная мутация, ген инактивируется путем метилирования; плод будет пораженным. Тяжесть заболевания зависит от степени Х-инактивации у женщин, степени метилирования и мозаицизма размера повторов.
Женщины-носители премутации имеют 50%-й риск передачи гена с экспансией. Мужчины с премутациею фенотипически являются нормальными, но все их дочери будут носителями премутации. В случае трансмиссии мужчинам количество повторов остается стабильным. Тест на ломку Х-хромосому выполняется с целью выявления количества повторов и степени метилирования.
Диагностика
Диагностику синдрома Эдвардса можно осуществить во время беременности Наиболее доступной методикой является ультразвуковое исследование.
Косвенные признаки синдрома Эдвардса на УЗИ на раннем этапе (12 недель):
- большой объем околоплодной жидкости;
- снижение частоты сердцебиения;
- брюшная грыжа;
- отсутствие носовых костей;
- 2 артерии в пуповине;
- кисты сосудистых сплетений.
На поздних сроках (третий триместр беременности) синдром Эдвардса на УЗИ может проявляться такими симптомами, как:
- аномалии костей головы и мягких тканей;
- пороки опорно-двигательного аппарата, сердца, сосудов, мочеполовой системы и так далее.
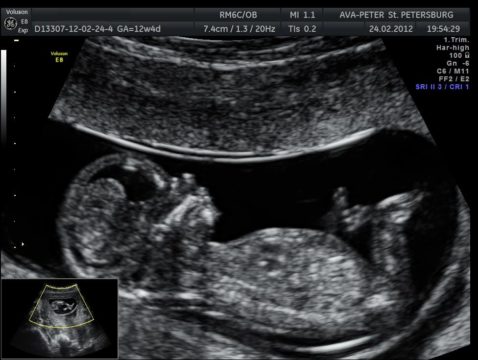 УЗИ – один из методов, позволяющих предположить синдром Эдвардса
УЗИ – один из методов, позволяющих предположить синдром Эдвардса
Более точным способом диагностики является биохимический анализ крови на 11-13 неделе беременности. Исследование предполагает определение уровня плазменного протеина А (РАРР-А) и хорионического гормона (β-ХГЧ). Затем полученные результаты соотносят с возрастом будущей мамы и сроком вынашивания ребенка. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. Диагноз на этом этапе не определяется.
Затем осуществляется определение статуса плода с помощью различных методик:
Инвазивные способы выявления синдрома Эдвардса:
- Биопсия ворсин хориона (БВХ) – исследование небольшого образца плаценты. Доступна на сроке 8-12 недель.
- Амниоцентез – проникновение через амниотическую оболочку и взятие пробы околоплодных вод. Делается на сроке 14-18 недель.
- Кордоцентез – забор пуповинной крови. Проводится в 20 недель.
Все три метода дают возможность определить кариотип ребенка и подтвердить либо опровергнуть диагноз. Их недостаток – риск инфицирования плода и прерывания беременности.
К неинвазивным методикам относится выделение ДНК ребенка из крови матери.
Точность генетического тестирования очень высока: синдром Эдвардса подтверждается часто – в 90% случаев. Сложнее всего установить мозаичную форму заболевания: в проверяемый образец могут не попасть клетки с измененным кариотипом.
После рождения ребенка патология диагностируется на основании внешнего осмотра и кариотипирования – исследования крови с целью определения хромосомного набора.
Причины
Причина синдрома Эдвардса – присутствие в наборе хромосом (кариотипе) «лишней» 18 хромосомы. В норме зигота (яйцеклетка после оплодотворения) должна иметь 23 пары хромосом, полученных в результате слияния женской и мужской гамет (половых клеток). Под влиянием не установленных наукой факторов происходит дублирование генетического материала. У зиготы вместо двух 18 хромосом появляется три: материнская, отцовская и дополнительная.
Мутация может осуществиться как до оплодотворения, так и после него – на этапе деления яйцеклетки. Кариотип при синдроме Эдвардса имеет три варианта:
- полная трисомия (95% случаев) – три 18 хромосомы в каждой клетке плода;
- транслокационная (2%) – две 18 хромосомы и часть третьей, добавившаяся к другой хромосоме;
- мозаичная (3%) – три 18 хромосомы в некоторых клетках будущего ребенка.
Первая форма трисомии синдрома Эдвардса характеризуется более сложным течением, чем две другие.
Общая информация
Трисомия по 18 хромосоме (синдром Эдвардса) – ненаследственное заболевание, мутация генов происходит случайно. Патология встречается в 1 случае из 3000 зачатий и 6000-7000 рождений. У девочек она диагностируется чаще, чем у мальчиков в 3 раза.
Риск синдрома Эдвардса повышается с возрастом матери – после 45 лет он составляет 0,7%. Болезнь негативно отражается на течении беременности: отмечается многоводие, недостаточная активность плода и роды позже срока (на 42-43 неделе).
60% детей с синдромом Эдвардса умирают в период внутриутробного развития. Продолжительность жизни 2-3 месяца – для мальчиков, 10 месяцев – для девочек. Смерть наступает из-за серьезных нарушений в работе внутренних органов.
Симптомы
Фото новорожденных c синдромом Эдвардса
Синдром Эдвардса проявляется у плода еще в период внутриутробного развития. В этот период признаки такой патологии могут быть обнаружены в процессе ультразвукового исследования. В ходе такого исследования может быть обнаружена их часть. 9 месяцев беременности – период, на протяжении которого женщина должна проходить УЗИ несколько раз. О синдроме Эдвардса могут свидетельствовать следующие симптомы:
- Существенная задержка внутриутробного развития – она может составлять несколько недель.
- Многоводие.
- Пороки развития головного мозга.
- Одна пупочная артерия у плода.
- Очень слабые шевеления плода.
- Большая длительность беременности – до 42 недель. При этом ребенок рождается с очень маленьким весом.
Признаки синдрома Эдвардса у новорожденных заметны сразу после рождения малыша. Отмечаются такие характерные симптомы:
- Низкая масса тела.
- Низкий лоб, широкий затылок.
- Долихоцефалический череп.
- Ушные раковины недоразвиты, расположены очень низко.
- Короткая верхняя губа, маленький рот, высокое небо.
- «Заячья» губа, «волчья» пасть.
- Патологические изменения опорно-двигательного аппарата – узкий таз, вывихнутые бедра, слишком широкая грудная клетка, косолапость.
- Сросшиеся пальцы на ногах, также они могут быть с перепонками.
- Неровное расположение пальцев.
- Искаженная форма лица.
- Очень маленькая голова.
Отдельно выделяется ряд симптомов, связанных с поражением внутренних органов, а также с нарушением нервно-психического развития:
Как правило, умственное развитие детей с таким заболеванием соответствует тяжелой степени олигофрении. Однако при мозаичной форме синдрома определенные достижения в умственном развитии у пациентов могут быть.
Частота появления
Риск рождения ребенка с трисомией 18 сильно зависит от возраста матери. Для восемнадцатилетней женщины риск рождения ребенка с трисомией 18 в утробе матери на 12-й неделе беременности составляет примерно 1: 2500. Этот риск возрастает примерно до 1: 180 для сорокалетней женщины. Поскольку около 80 процентов детей с трисомией 18 умирают в утробе матери между 12-й неделей беременности и родами, вероятность рождения ребенка соответственно в 5-6 раз ниже, чем указано для 12-й недели беременности. Это делает синдром Эдвардса вторым по распространенности заболеванием, вызванным трисомией среди живорожденных детей, после синдрома Дауна ( трисомия 21 ).
Девочки страдают значительно чаще, чем мальчики: из 100 детей с синдромом Эдвардса в среднем 75 — девочки и 25 — мальчики ( гинекотропия / соотношение f: m = 3: 1).
Пренатальная диагностика
1) Инвазивные исследования (амниоцентез, биопсия хориона) в основном назначают тем женщинам, у которых наблюдается повышенный риск того, что родится малыша с синдромом Эдвардса, например, пациенткам, чей возраст превышает 35 лет или с плохими результатами неинвазивных тестов: УЗИ и анализов. Инвазивные методы диагностики являются высокоточными, однако, учитывая риск осложнений, не подходят для массового проведения всем беременным, а проводятся только по особым показаниям.
2) Неинвазивные технологии, так называемые скрининги. Скрининг – комплексное исследование беременных женщин на наличие у плода хромосомных аномалий. Выделено несколько признаков, указывающих на высокий риск наличия заболевания, которые может выявить УЗИ плода (отсутствие носовой кости, увеличенная толщина воротникового пространства, недостаточная длина бедренных и плечевых костей и другие особенности). В комплексе с УЗИ идёт биохимический анализ крови матери на такие гормоны как свободный бета-ХГЧ и PAPP-A. Полученные данные по биохимическим маркерам анализируют в совокупности с результатами ультразвукового исследования, а результат всего скрининга представляет собой расчет риска наличия хромосомной аномалии у плода.
Однако при использовании стандартных тестов на синдром Эдвардса, лишь у 3% женщин, направленных на инвазивную диагностику действительно подтверждается наличие заболевания. В то же время не исключены и ложно-отрицательные результаты, когда скрининг показывает низкий риск, а ребенок рождается с хромосомной патологией.
- точность 99%, что намного точнее классической диагностики (УЗИ и биохимический скрининг)
- совершенно безопасен, в отличие от инвазивных методик — для забора материала на анализ необходимо просто взять кровь из вены беременной женщины.
- на ранних сроках: анализ можно проводить уже на 9-й неделе беременности.
Что после рождения?
Младенцы, рожденные с синдромом Эдвардса, обычно рождаются недоношенными, с очень низкой массой тела при рождении и требуют интенсивной терапии с самого рождения. Дефекты, связанные с возникновением этого заболевания, могут включать многие системы и органы, в том числе: пороки сердца, гортань, диафрагмальную грыжу, стриктуру пищевода, гидронефроз и грыжу позвоночника.
Выживаемость новорожденных с синдромом Эдвардса составляет около 50% в первые недели жизни. Однако до 90% детей умирают в возрасте до 1 года.
Обычно кормление ребенка необходимо через зонд, введенный в желудок или энтерально, из-за нарушения сосательного и глотательного рефлексов. Малыши также находятся под наблюдением кардиолога, невролога, нефролога, генетика, а также нуждаются в реабилитации для улучшения работы органов движения.
Синдром Эдвардса
Синдром Эдвардса характеризуется трисомией по 18 хромосоме и комплексом множественных пороков развития.
В одном случае из 10 наблюдается мозаицизм, то есть лишняя хромосома есть не во всех клетках организма. Возможна и частичная трисомия с присоединением части 18 хромосомы к другой хромосоме.
Во время беременности наблюдается малый вес плода, многоводие, небольшая плацента и наличие одной артерии плаценты.
Новорожденные имеют изменение формы черепа, маленькие рот и целюсть, лицевой дисфорфизм, дефекты глаз и низкие деформированные ушные раковины. Также наблюдаются численные аномалии пальцев рук и ног, деформация стопы («стопа-качалка»).
Из дефектов внутренних органов наиболее часто встречаются пороки сердца и сосудов. У всех наблюдается гипоплазия мозжечка.
Синдром Эдвардса характеризуется умственной отсталостью и задержкой в развитии.
Большая часть детей умирает в первые месяцы жизни.
Симптомы
Синдром Эдвардса у детей проявляется наличием таких признаков:
- нарушения в развитии опорно-двигательного аппарата и костей черепа;
- нарушения в развитии нижней челюсти ребенка;
- недоразвитие ротового отверстия;
- удлинение черепа в переднем или заднем направлении;
- глазные щели – узкие;
- сращение пальцев (бывает частичное или полное) стоп и кистей;
- изменения в форме стоп, а также запястных суставов;
- редко могут возникнуть расщелины губы или неба;
- нарушение в развитии скелетной мускулатуры;
- нарушения в развитии подкожной клетчатки.
Со стороны нервной системы могут возникать такие нарушения как недоразвитость головного мозга, задержка в психическом или же физическом развитиях. Могут также возникнуть различные пороки внутренних органов, такие как:
- дефекты сердечных клапанов;
- дефекты сердечных перегородок;
- изменения в крупных сосудах;
- аномалии в развитии пищевода;
- нарушения в работе мочеполовой системы (удвоение почек, расширения отделов мочеполовой системы);
- нарушения в развитии половых органов;
- неправильное расположение в организме органов брюшной полости;
- заращение желчевыводящей системы.
Если смотреть со стороны зрения, то возможно уменьшение глазных щелей.
Трисомии
В материале выкидышей трисомии представляют более половины всех количественных хромосомных аберраций
Обращает на себя внимание то, что в случаях моносомий недостающей хромосомой обычно оказывается X-хромосома, а в случаях избыточных хромосом, дополнительная хромосома чаще всего оказывается аутосомой
Точная идентификация дополнительной хромосомы стала возможна благодаря методу G-бэндинга. Исследования показали, что все аутосомы могут принимать участие в нон-дисджанкшн (см. таблицу)
Обращает на себя внимание, что три хромосомы, чаще всего встречающиеся при трисомиях новорожденных (15-я, 18-я и 21-я) чаще всего обнаруживаются и при летальных трисомиях у зародышей. Вариации относительных частот различных трисомий у зародышей отражают во многом сроки, на которых происходит гибель зародышей, поскольку, чем более летальной является комбинация хромосом, тем на более ранних сроках происходит остановка развития, тем реже будет обнаруживаться такая аберрация в материалах выкидышей (чем меньше срок остановки развития, тем труднее обнаружить такой зародыш)
| Лишняя хромосома при летальных трисомиях у зародыша (данные 7 исследований: Буэ (Франция), Карр (Канада), Кризи (Великобритания), Дилл (Канада), Кадзи (Швейцария), Такахара (Япония), Теркелсен (Дания)) | ||
|---|---|---|
| Дополнительная аутосома | Количество наблюдений | |
| A | 1 | |
| 2 | 15 | |
| 3 | 5 | |
| B | 4 | 7 |
| 5 | ||
| C | 6 | 1 |
| 7 | 19 | |
| 8 | 17 | |
| 9 | 15 | |
| 10 | 11 | |
| 11 | 1 | |
| 12 | 3 | |
| D | 13 | 15 |
| 14 | 36 | |
| 15 | 35 | |
| E | 16 | 128 |
| 17 | 1 | |
| 18 | 24 | |
| F | 19 | 1 |
| 20 | 5 | |
| G | 21 | 38 |
| 22 | 47 |
Пренатальная диагностика
1) Инвазивные исследования (амниоцентез, биопсия хориона) в основном назначают тем женщинам, у которых наблюдается повышенный риск того, что родится малыша с синдромом Эдвардса, например, пациенткам, чей возраст превышает 35 лет или с плохими результатами неинвазивных тестов: УЗИ и анализов. Инвазивные методы диагностики являются высокоточными, однако, учитывая риск осложнений, не подходят для массового проведения всем беременным, а проводятся только по особым показаниям.
2) Неинвазивные технологии, так называемые скрининги. Скрининг – комплексное исследование беременных женщин на наличие у плода хромосомных аномалий. Выделено несколько признаков, указывающих на высокий риск наличия заболевания, которые может выявить УЗИ плода (отсутствие носовой кости, увеличенная толщина воротникового пространства, недостаточная длина бедренных и плечевых костей и другие особенности). В комплексе с УЗИ идёт биохимический анализ крови матери на такие гормоны как свободный бета-ХГЧ и PAPP-A. Полученные данные по биохимическим маркерам анализируют в совокупности с результатами ультразвукового исследования, а результат всего скрининга представляет собой расчет риска наличия хромосомной аномалии у плода.
Однако при использовании стандартных тестов на синдром Эдвардса, лишь у 3% женщин, направленных на инвазивную диагностику действительно подтверждается наличие заболевания. В то же время не исключены и ложно-отрицательные результаты, когда скрининг показывает низкий риск, а ребенок рождается с хромосомной патологией.
- точность 99%, что намного точнее классической диагностики (УЗИ и биохимический скрининг)
- совершенно безопасен, в отличие от инвазивных методик — для забора материала на анализ необходимо просто взять кровь из вены беременной женщины.
- на ранних сроках: анализ можно проводить уже на 9-й неделе беременности.
Лечение
Коррекция патологических процессов, опасных для жизни, заключается в назначении противовоспалительного и антибактериального лечения при наличии пневмонии, восстановлении проходимости пищеварительного тракта при атрезии кишечника, кормлении через зонд, использовании пеногасителей при метеоризме, слабительных средств при запоре. Для поддержания жизнеспособности всего организма проводится коррекция работы дыхательной и пищеварительной систем, а также нормализация сердечной деятельности. Больным необходимо создать стерильные условия, чтобы избежать развития различных инфекционных заболеваний. Дети с данным синдромом требуют тщательного ухода и регулярного прохождения медицинского обследования.

Пупочные и паховые грыжи, пороки сердца и прочие аномалии лечат хирургическим путем, если состояние больного ребенка остается удовлетворительным. Оперативное вмешательство несет неоправданный риск. Хирурги, исправив внешние дефекты, могут потерять пациента из-за сбоя в работе сердечно-сосудистой системы и развития прочих серьезных осложнений.
Все дети с синдромом Эдвардса без исключения имеют отклонения в психофизическом развитии. Им требуется специальная программа воспитания, включающая комплекс обучающих методик. Она не восстановит нарушенные функции, но поможет привить некоторые элементарные бытовые навыки.
Диагностика после рождения
Синдром Эдвардса у новорожденных обнаруживают по таким признакам:
- небольшой вес новорожденного
- микроцефалия
- волчья пасть или заячья губа
- поперечная ладонная борозда
- неразвитая на пальцах дистальная сгибательная складка
- дистальное расположение осевого трирадиуса и увеличение гребневого счета на ладони
- дуги на подушечках пальцев рук
Но эти признаки еще не говорят о том, что у ребенка именно синдром Эдвардса. Диагноз нуждается в подтверждении. Для этого применяют выше упомянутый метод КФ-полиплазменной цепной реакции, чтобы определить каропин новорожденного. УЗИ показывает кисты сосудистых сплетений у младенца.
Моносомии
Моносомия X (45,X) представляет одну из часто встречающихся аномалий в материале самопроизвольных выкидышей. При рождении она соответствует синдрому Шерешевского-Тернера, и при рождении она встречается реже, чем другие количественные аномалии половых хромосом. Эта бросающаяся в глаза разница между относительно высокой частотой обнаружения лишних X-хромосом у новорожденных и относительно редким обнаружением моносомии X у новорожденных указывает на высокую частоту летальности моносомии X у зародыша
Кроме того, обращает на себя внимание очень большая частота мозаик у больных с синдромом Шерешевского-Тернера. В материале выкидышей, наоборот, мозаики с моносомией X крайне редки
Данные исследований показали, что только менее 1% всех моносомий X доходит до срока родов. Моносомии аутосом в материале выкидышей встречаются довольно редко. Это очень контрастирует с высокой частотой соответствующих трисомий.
Диагностика
О генетических патологиях чаще всего можно узнать, пока женщина еще вынашивает ребенка. Это касается и трисомий. Скрининг беременности проводят с 11-й по 13-ю неделю. Женщина сдает анализы крови (биохимию), проводится УЗИ. Также диагностика заключается в определении каротипа эмбриона, если женщина находится в группе риска (отягощенный семейный анамнез, инфекционные болезни в первом триместре и т. д.).
В скрининге первого семестра определяют, сколько в крови хорионического гормона человека и плазменного протеина А ассоциированного с беременностью. Потом учитывают возраст беременной, чтобы узнать, с каким риском у нее может родиться ребенок с трисомией 18.
Если женщину отнесли в группу риска, чуть позже делают биопсию плода, чтобы точно знать, родится ли ребенок с отклонениями, или здоровый. С 8 до 12 неделю берется анализ ворсин хориона. С 14 по 18-ю неделю проводится изучение вод, окружающих плод. После 20-й недели могут сделать кордоцентез. Процедура подразумевает, что возьмут крови из пуповины (в процессе применяется ультразвук для контроля взятия материала).
В материале обнаруживают количество хромосом. В этом помогает метод КФ–ПЦР. При условии непрохождения беременной генетического скрининга на поздник сроках гестации делают предварительную диагностику генетической мутации методом ультразвукового исследования. Во втором и третьем триместре есть признаки, которые говорят о том, что ребенок с большой вероятностью родится с трисомией:
- заячья губа
- низко расположенные уши плода
- микроцефалия
- волчья пасть
- пороки опорно–двигательного аппарата
- пороки развития мочеполовой системы
- пороки сердца и сосудов
Что такое ранний пренатальный скрининг и когда он проводится
Скрининг (от англ. «просеивание») – это совокупность исследований, позволяющих определить группы беременных, у которых существует риск рождения ребенка с хромосомными аномалиями и врожденными пороками. Но ранний скрининг – это только начальный, предварительный этап обследования, после которого женщинам, с выявленным риском врожденных аномалий, рекомендуется более детальное диагностическое обследование, которое точно подтвердит или исключит наличие патологии.
Данный комплекс диагностики проводится беременным на сроке 11–13 недель +6 дней (когда копчико-теменной размер плода составляет от 45 до 84 мм). Самым оптимальным временем для проведения скрининга считается возраст от 12 до 13 недели, так как при таком обследовании оцениваются как признаки хромосомной патологии у плода, так и анатомические структуры будущего ребенка, а на сроке в 11 недель – 11 недель +4 дня сделать это очень сложно.
Что включает в себя ранний пренатальный скрининг
В комплексное обследование входят:
- УЗИ плода на наличие маркеров (признаков) хромосомной патологии и врожденных пороков развития;
- определение уровня биохимических маркеров хромосомных аномалий в крови беременной женщины: бета-ХГЧ и РАРР-А. Исследование проходит на специальном высокотехнологичном оборудовании Cobas e, Roshe, разрешенном для расчета риска хромосомных синдромов в программе Astraia (FMF);
- допплерометрия маточных артерий;
- цервикометрия – измерение длины шейки матки;
- сбор анамнеза (возраст, рост, вес, количество беременностей и родов, курение, сахарный диабет, артериальная гипертензия и др.);
- измерение артериального давления на обеих руках.
Полученные данные: анамнез, УЗИ и биохимические маркеры помещают в специально разработанную программу Astraia, которая рассчитывает риск рождения ребенка с врожденными аномалиями. Комбинация данных исследований увеличивает эффективность выявления плодов с синдромом Дауна и другими хромосомными заболеваниями.
Показания для тестирования на ломкую Х-хромосому
- Индивиды с задержкой умственного и общего развития, аутизмом
- Индивиды с чертами фрагильной Х-хромосомы
- Индивиды с наличием синдрома фрагильной Х-хромосомы в семейном анамнезе
- Индивиды с наличием в семейном анамнезе недиагностированной задержки умственного развития
- Плоды от матерей-носителей
Геномный импринтинг — процесс, при котором активация гена происходит преимущественно в материнской или преимущественно в родительской хромосоме, но не в обеих хромосомах. Нормальное развитие имеет место лишь в том случае, если присутствуют обе копии (материнская и отцовская) импринтинг-ген. Импринтинг-ген неактивен, значит, активный ген теряет (путем делеции) или получает мутацию, в таком случае плод будет пораженным. Лишь несколько генов могут испытывать импринтинга. Примерами геномного импринтинга может быть синдром Ангельмана и полный пузырный занос (вариант гестационной трофобластической болезни).
Синдром Ангельмана характеризуется тяжелой задержкой умственного развития, атаксической походкой, типичным лицом, пароксизмами смеха и судорогами. Ген синдрома Ангельмана является активным только в материнской унаследованной хромосоме, следовательно, если происходит делеция материнской хромосомы 15 или материнская копия гена имеет мутацию, белковый продукт не образуется и плод будет пораженным.
Синдром Ангельмана также может развиться, если обе копии хромосомы 15 является унаследованными от отца (отсутствие материнской копии хромосомы 15). Это состояние получило название унипарентальной дисомии. Унипарентальная дисомия возникает чаще вследствие потери хромосомы у эмбриона с трисомией или добавления хромосомы у плода с моносомией по этой хромосомой. Каждая из хромосом может быть генетически различной (гетеродисомия) или идентичной (изодисомия), в зависимости от времени возникновения этого феномена — в течение первого или второго мейотического деления, соответственно.
Полный пузырный занос обычно является диплоидным (46, ХХ или Х ¥), но может иметь полностью отцовское происхождение, без материнского хромосомного материала. При таких условиях плод не может развиваться. Полный пузырный занос может сопровождать нормальную многоплодную беременность, но в этом случае возрастает риск материнских осложнений (гипертиреоидизм, преэклампсия, преждевременные роды). В отличие от полного пузырного заноса, частичный пузырный занос обычно является триплоидным (69, ХХХ, 69, ХVV), с дополнительным набором отцовских хромосом.
Триплоидия с дополнительным набором материнских хромосом имеет место при ЗВУР плода, врожденных пороках развития и маленькой плаценте.