Синдром прадера-вилли как генетическая патология
Содержание:
Общие сведения о синдроме Вилли-Прадера
Первое упоминание о патологии датируется 1887 годом. Лэнгдон Даун описал девочку-подростка, у которой отмечалась задержка физического развития, гипогонадизм и ожирение. Первоначально болезнь получила название «полисарция». Полноценную характеристику синдрому дали швейцарские врачи Прадер, Вилли и Лабхарт в 1956 году. Позднее в ходе глубокого изучения, доктора определили и точную локализацию генетической мутации, которая приводила к возникновению заболевания у детей. Они также связали изменения с синдромом Ангельмана. Оба расстройства провоцируются дефектом в строении 15 хромосомы. При этом в одном случае аномалия формируется в материнской копии, а в другом – в отцовской. Патология была названа синдромом Вилли-Прадера в честь врачей, внесших самый большой вклад в ее изучение. Болезнь относится к числу редких, так как ее распространенность колеблется в пределах одного случая на 10–25 тысяч новорожденных. Половой или расовой предрасположенности не установлено.
Профилактика
Предотвратить врожденное заболевание невозможно, главное в этом случае – не допустить появления осложнений. Лечение синдрома следует начать как можно раньше, тогда ребенку будет проще приспособиться к обучению в школе и к жизни в обществе.
К профилактике заболевания можно отнести медико-генетические консультации семей, у которых есть предрасположенность к возникновению синдрома. Будущим родителям необходимо провести дородовое генетическое исследование, которое поможет определить особенности строения хромосом плода.
Чтобы улучшить жизнь ребенка с СПВ, следует обеспечить постоянное сотрудничество специалистов медицинских учреждений, родителей и самого малыша.
Диагностика СПВ
Диагностика болезни на начальных ее стадиях позволяет предотвратить развитие некоторых ее симптомов:
- Терапия, начатая на ранней стадии, вырабатывает у ребенка правильное пищевое поведение;
- Если до 18 месяца жизни специалисты стали корректировать соотношение гормонов роста, телосложение малыша будет развиваться правильно, как и у здорового человека.
Большие признаки, равные одному баллу:
- Периодические трудности с кормлением новорожденного;
- Задержка в познавательном развитии до 5-6 лет;
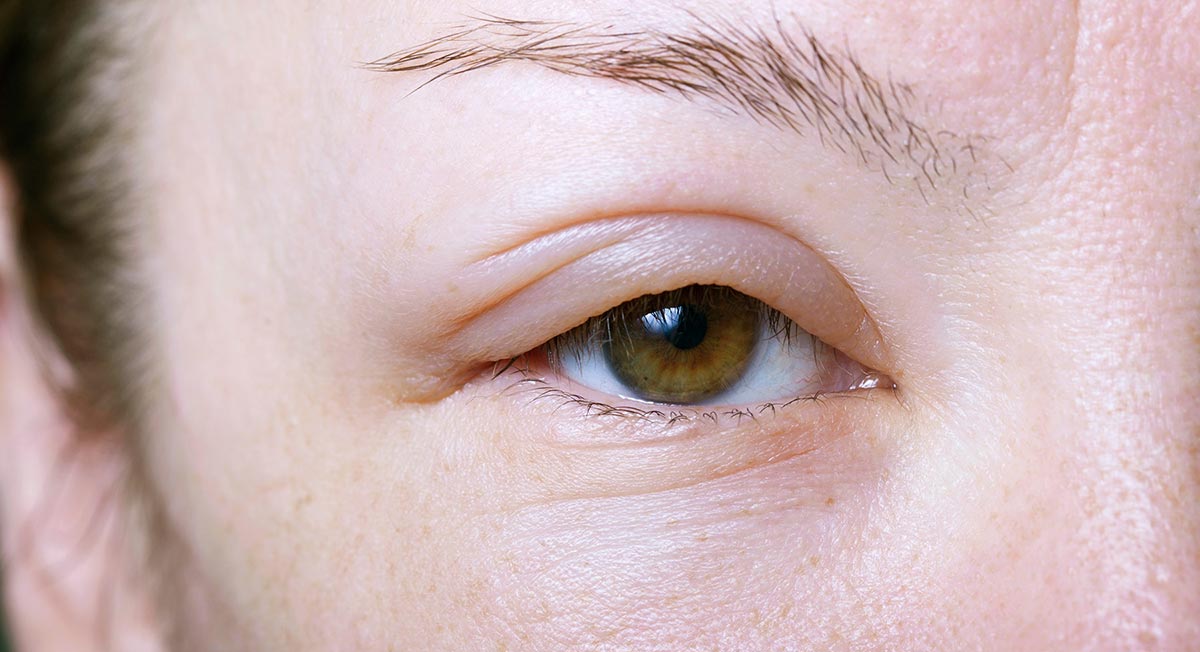
- Особые черты лица: миндалевидный разрез глаз, небольшой рот, узкая верхняя губа;
- Гипотония мышц, выявленная еще в возрасте от 1 до 3 лет;
- Изменения в строении органов репродуктивной системы;
- Развитие ожирения.
Малые признаки (0,5 балла):
- Недостаточная активность плода;
- Аномалии рефракции;
- Поражение кожных покровов;
- Пониженная пигментация радужки глаза, волос и кожного покрова;
- Густая слюна;
- Невысокий рост;
- Непропорциональные конечности;
- Проблемы со сном;
- Психические отклонения в поведении;
- Нарушение артикуляции.
Помимо вышеперечисленных критериев для точного определения диагноза следует провести кариотипирование и определить наличие различных модификаций на уровне 15 хромосомы. Также используют ДНК-маркеры и метод прометафазного анализа.
Часто патология становится заметной уже во время ультразвукового исследования беременности. Специалист замечает увеличение околоплодных вод, гипоксию плода или его нестандартное расположение. При малейших подозрениях на наличие нарушения будущей маме придется пройти перинатальную диагностику, включающую генетическое тестирование и анализ крови на уровень гонадотропина. Также для определения синдрома необходимо применить специальные молекулярно-генетические маркеры.
Дети, больные СПВ, мало двигаются, нередко крадут продукты, прячут еду и, даже несмотря на недавний перекус, постоянно остаются голодными. В таком случае появляется угроза возникновения апноэ – остановки дыхания во сне, опасной возможным смертельным исходом.
Профилактика
Предотвратить врожденное заболевание невозможно, главное в этом случае – не допустить появления осложнений. Лечение синдрома следует начать как можно раньше, тогда ребенку будет проще приспособиться к обучению в школе и к жизни в обществе.
К профилактике заболевания можно отнести медико-генетические консультации семей, у которых есть предрасположенность к возникновению синдрома. Будущим родителям необходимо провести дородовое генетическое исследование, которое поможет определить особенности строения хромосом плода.
Чтобы улучшить жизнь ребенка с СПВ, следует обеспечить постоянное сотрудничество специалистов медицинских учреждений, родителей и самого малыша.
Не только редкие, но и «прячущиеся»
Одна из сложностей диагностирования некоторых орфанных заболеваний заключается в большой схожести между их симптомами и симптоматикой других патологий. Например, орфанные заболевания могут «маскироваться» под:
- ДЦП
- Ранний детский аутизм
- Спинальную мышечную атрофию
И другие распространенные заболевания.
В этом случае знаний обычного невролога, педиатра или врача другого профиля явно недостаточно, ребенка должен осмотреть опытный специалист именно по редким генетическим заболеваниям у детей. Обратившись в Хадассу, вы получите консультацию и лечение у ведущих экспертов в данной области и сможете быть полностью уверенными в правильности диагноза и максимальной эффективности назначенной терапии.
Лечение
Синдром Прадера-Вилли, проявляющийся у детей, до сегодняшнего дня не имеет специального лечения. При обнаружении нарушений дыхательного рефлекса, новорожденные подключаются к аппарату ИВЛ. Нарушение глотательного рефлекса и питания на первых порах устраняют с помощью зонда. Пониженный тонус мышц предусматривает проведение массажа и физиотерапевтических процедур.
Детям вводится гормон роста, который сопутствует процессу увеличения мышечной массы и снижению аппетита. Нарушения в мочеполовой системе предусматривает введение больным детям специального гормона, применение которого направлено на своевременное половое созревание. В случае неопущенности яичек ребенок становится на учет к детскому андрологу. Эту проблему решают и хирургическим путем, если яички со временем не опустились до нормального состояния.
В некоторых случаях необходима и консультация психиатра. Задержка развития речевого аппарата и психологического состояния требуют помощи психолога. Родителям следует серьезно отнестись к рациону ребенка, защитить его от переедания, ввести в употребление те продукты, которые посоветует диетолог
Следует уделить огромное внимание социализации ребенка, дать ему шанс на занятие спортом, общение со сверстниками. Появление второго ребенка в семье следует планировать, пройти генетическую экспертизу, так как риск рождения еще одного ребенка с патологией достаточно высок
Детям необходима постоянная консультация эндокринолога и невропатолога.
Улучшение общего состояния
Синдром Прадера-Вилли сопровождается соматическими осложнениями, трудностями в общении, поэтому необходима специальная медицинская помощь и подход к носителям данной болезни.
Они, как правило, не имеют представление о сохранности своего здоровья, что усугубляет общее состояние и соматические патологии. Хорошее состояние здоровья должно стать основной движущей силой у таких людей.
Они мало проявляют способности к обучению, часто меняют свои приоритеты в жизни. Очень важна для таких людей среда, социализация в ней, чтобы они стали членами общества.
Генетика
PWS связан с эпигенетическим феноменом, известным как импринтинг . Обычно плод наследует импринтированную материнскую копию генов PW и функциональную отцовскую копию генов PW. Из-за импринтинга унаследованные от матери копии этих генов практически не работают, и поэтому плод полагается на экспрессию отцовских копий генов. Однако при PWS происходит мутация / делеция отцовских копий генов PW, в результате чего у плода отсутствуют функционирующие гены PW. Гены PW являются SNRPN и NDN necdin гены, наряду с группами snoRNAs : SNORD64 , SNORD107, SNORD108 и две копии SNORD109, 29 копий SNORD116 (HBII-85) и 48 копий SNORD115 (HBII-52). Эти гены расположены на хромосоме 15, расположенной в районе 15q11-13. Эта так называемая область PWS / AS в отцовской хромосоме 15 может быть потеряна одним из нескольких генетических механизмов, что в большинстве случаев происходит в результате случайной мутации. Другие, менее распространенные механизмы включают дисомию у одного родителя , спорадические мутации , транслокации хромосом и делеции генов.
Область 15q11-13 участвует как в PWS, так и в синдроме Ангельмана (AS). В то время как PWS является результатом потери генов PW в этой области на отцовской хромосоме, потеря другого гена ( UBE3A ) в той же области на материнской хромосоме вызывает AS. PWS и AS представляют собой первые зарегистрированные случаи расстройств, связанных с импринтингом у людей.
Риск СПВ для брата или сестры пострадавшего ребенка зависит от генетического механизма, вызвавшего заболевание. Риск для братьев и сестер составляет <1%, если пораженный ребенок имеет делецию гена или однопородную дисомию, до 50%, если пораженный ребенок имеет мутацию контролирующей области импринтинга, и до 25%, если присутствует родительская хромосомная транслокация. Пренатальное тестирование возможно на любой из известных генетических механизмов.
Микроделеционный в одной семье snoRNA HBII-52 исключила его из играет важную роль в развитии этого заболевания.
Исследования модельных систем человека и мыши показали, что основной причиной СПВ является делеция 29 копий C / D box snoRNA SNORD116 (HBII-85).
Эндокринные нарушения
Существует несколько факторов, подтверждающих концепцию дефицита роста у лиц подверженных синдрому.
- Лица, подверженные болезни, имеют низкий рост и страдают от чрезмерного ожирения, то есть у них пониженное содержание свободной жировой массы, пониженная плотность костной ткани, низкий уровень использованной энергии.
- Для данной болезни характерны нарушения в мочеполовой системе. У мужского пола проблемы возникают с неопущением яичек (со временем яички могут опуститься до нормального уровня), а у женского в появлении адренархе. В обоих случаях возможно решение проблемы хирургическим путем.
Трудно ли было смириться с тем, что ваш ребёнок болен?
Марина А.: Два года назад, когда я узнала о синдроме, написала текст для поста в инстаграме, но опубликовала его только недавно (Марина открыто рассказала о заболевании сына на аудиторию в 88 тысяч человек. — Примеч. ред.). У меня не было проблем с принятием. Конечно, потребовалось время, чтобы собраться, но я быстро взяла себя в руки и поняла, что раз в моей жизни произошла такая ситуация, то надо сделать что-то полезное. Я решила использовать свои ресурсы, чтобы донести до людей информацию и помочь тем, кому это необходимо. Пришлось немного подождать, пока остальные члены семьи тоже будут готовы говорить, и эта готовность совпала с открытием фонда, поэтому решила приурочить.
- Марина Вербивская с дочкой Кристиной
- Марина Амулина с сыновьями Леоном и Робертом
Марина В.: Мой момент принятия начался ещё во время беременности, когда я поняла, что есть какие-то сложности. Как и у всех, когда узнала диагноз, был шок. Месяц в слезах. Я не могла брать ребёнка на руки, потому что она была слишком слаба, лежала под трубками. Но я знала, что когда мы приедем домой, там нас будут ждать и любить несмотря ни на что пять членов нашей семьи, мои родители и родители мужа. Эта любовь важнее, чем какое-то отклонения от нормы.
Что такое микроделеционный синдром?
Самые незначительные изменения (они же мутации) называются точечными. Их появление влияет на считанные единицы генов. В некоторых случаях нарушение относится вообще к одному единственному гену
Однако если он обеспечивал выработку важного белка, последствия для всего организма могут быть очень серьезными. Подобные патологические изменения относятся к группе микроделеционных синдромов
Каждое такое заболевание обусловлено небольшим изменением генетического материала, которое происходит в строго определенном месте. Точный механизм возникновения подобных нарушений на сегодняшний день не установлен, что не мешает ученым заниматься исследованием их воздействия на организм.
Так, было выяснено, что развитие синдрома в таком случае может происходить несколькими различными вариантами. В частности, ряд заболеваний характеризуется участием онкогенов. В других случаях на воздействие непосредственно самой делеции накладывается эффект хромосомного импринтинга и возможные однородительские дисомии.
Частота возникновения большей части микроделеционных синдромов крайне невелика: порядка 1 случая на 50-100 тысяч новорожденных. Набор клинических признаков обычно выражается отчетливо. Для того чтобы поставить диагноз, бывает достаточно лишь совокупности симптомов. Однако при таком подходе невозможно точно прогнозировать здоровье потомков, поэтому зачастую наряду с проверкой обычных признаков производится молекулярно-генетическая диагностика пробанда и его родственников (обычно родители, в некоторых случаях также требуется анализ генотипа братьев, сестер, теть, дядь и так далее).
Патологические проявления сильно отличаются. В частности, их проявление определяется тем, насколько большой участок генетического материала был утрачен в результате делеции. Кроме того, в ряде случаев играет роль то, от кого из родителей была получена мутация (влияние хромосомного импринтинга). Хорошей иллюстрацией последней ситуации является пара синдромов Прадера-Вилли и Ангельмана. Они оба обусловлены наличием делеции в 15 хромосоме. Однако из-за различного механизма действия при передаче от разных родителей клиническая картина этих заболеваний значительно отличается.
Когда и почему возникают генетические патологии плода: риски по возрастам
Аномалии развития плода закладываются уже в момент оплодотворения сперматозоидом яйцеклетки. Например, такая патология, как триплоидия (наличие трех хромосомом в ряду цепочки, а не двух, как положено), возникает в случае проникновения в яйцеклетку двух сперматозоидов, каждый из которых оставляет по одной хромосоме. Естественно, с таким набором живой организм не может выжить, поэтому на определённом этапе происходит выкидыш или замершая беременность.
В целом хромосомные патологии разделяются на 4 группы:
- Гаметопатия. Патология имеется ещё до зачатия в самом сперматозоиде или яйцеклетке, т.е. это генетическое заболевание — врожденная патология.
- Бластопатия. Аномалии возникают в первую неделю развития зиготы.
- Эмбриопатия. Повреждения эмбрион получает в период от 14 до 75 дней после зачатия.
- Фетопатия. Заключается в формировании патологии развития плода начиная с 75 дня после оплодотворения.
Данные статистики наводит на мрачные мысли. Так, риск рождения малыша с хромосомными аномалиями у 20-летних женщин составляет 1:1667, а у 35-летних уже 1:192. А на деле это означает, что в 99,5% случаев ребёнок у тридцатипятилетней матери родится здоровым.
Что бы вы сказали семьям, которые столкнулись с синдромом?
Марина В.: Когда родители только узнают о диагнозе, они пытаются предугадать будущее: а как будет у нас, пойдём на поправку или состояние ухудшится? Мне очень хочется, чтобы люди не теряли время, приняли ситуацию и проводили время с детьми. Дети быстро растут, та любовь, которая есть здесь и сейчас, должна занимать сто процентов нашего времени и сто процентов нашего внимания.
Марина А.: Есть такая фраза: «То, что вы можете воспринимать спокойно, больше не управляет вами»
Я думаю, она применима в нашей ситуации: важно уделять время своим переживаниям, но не допускать, чтобы они управляли вами. . Я советую изучить информацию, проанализировать её и понять, что это не беда, не ужасная проблема, а обстоятельства
Они так сложились, и мы никак не можем повлиять на этот факт. Но мы можем крепко любить детей, можем прикладывать все усилия, чтобы у них была полноценная жизнь. Я верю, что прогресс малыша, несмотря на исходные данные, на 50% зависит от настроя родителей
Я советую изучить информацию, проанализировать её и понять, что это не беда, не ужасная проблема, а обстоятельства. Они так сложились, и мы никак не можем повлиять на этот факт. Но мы можем крепко любить детей, можем прикладывать все усилия, чтобы у них была полноценная жизнь. Я верю, что прогресс малыша, несмотря на исходные данные, на 50% зависит от настроя родителей.
Как вас поддерживают мужья?
Марина А.: Без моего мужа и его поддержки я бы не справилась. Когда я узнала о диагнозе, позвонила ему и плакала, а он сказал: «Я не понимаю, почему ты так реагируешь, ничего страшного не случилось, мы со всем справимся». В тот момент он придал мне невероятных сил. Мой муж — моя самая большая опора. И у нас очень большая и любящая семья, она дарит непоколебимое чувство заботы и защиты, бесконечной любви и нежности. Роберт это однозначно чувствует.
Марина В.: Мой муж помогает мне и психологически, и физически. Всегда готов подхватить. Он очень активный с детьми, и им не страшно ничего в этой жизни. Он не относится к Кристине как к какому-то нездоровому ребёнку — точно так же играет, щекочет, подбрасывает. Но мы понимаем, что есть семьи, у которых такого нет, и мужчины уходят. Мы хотим показывать положительный опыт, что можно жить ещё и так.
Аниридия
При аниридии нарушается нормальное строение глаза: в органе зрения отсутствует радужная оболочка. Кроме того, часто развиваются сопутствующие патологические изменения, такие как макулярная гипоплазия и гипоплазия зрительного нерва, изменения роговицы, катаракта. Острота зрения заметно падает, попытки коррекции не приносят существенных результатов. Развивается светобоязнь и горизонтальный нистагм. В некоторых случаях отмечается появление врожденной глаукомы.
Причиной заболевания является нарушение функционирования гена PAX6 из короткого плеча 11 хромосомы. Кодируемый им белок приводит к запуску ряда процессов, которые управляют процессом правильного формирования органов зрения и ряда других структур. Примечательно, что ген очень консервативен: отличие форм PAX6 у человека и данио рерио составляет менее 5%, несмотря на расхождение эволюционных линий примерно 400 млн лет назад.
Заболевание относится к группе аутосомно-доминантных патологий. В случае гомозиготности по мутантной копии гена PAX6 негативный эффект на организм возрастает, что вызывает множественные нарушения в работе органов зрения. Кроме того, поражается ЦНС, что приводит к летальному исходу.
Лечение направлено на сглаживание симптомов. Для визуальной имитации зрачка рекомендуется использовать специальным образом окрашенные линзы. Возможно восстановление зрачка путем реконструктивной пластической операции.
Разновидности УЗИ исследований
Ультразвуковая диагностика представляет широкий спектр исследований. Существует несколько видов УЗИ, которые с предельной точностью определяют внутриутробные пороки развития малыша.
Стандартное УЗИ. Оно обычно совмещено с биохимическим анализом крови. Оно проводиться не раньше 10 недель беременности. В первую очередь у плода выявляют толщину воротниковой зоны, которая не должна превышать 3 мм, а также визуализацию носовой кости. У малыша с синдромом Дауна воротниковая зона толще нормы, а носовые кости не развиты. Также на увеличение толщины влияют следующие факторы:
- порок сердца
- застой крови в шейных венах
- нарушение лимфодренажа
- анемия
- внутриутробные инфекции
- 3D УЗИ позволяет увидеть цветное изображение малыша, разглядеть конечности, отсутствие сросшихся пальчиков, недоразвитых стоп и пр. Точность диагностики воротникового пространства увеличивается на 30%. Врач может точно сказать, имеются ли патологии развития нервной трубки.
- 4D УЗИ по принципу работы не отличается от более простых вариантов, но обладает массой преимуществ. Врач видит трёхмерное изображение сердца, вид плода с разных ракурсов. Именно 4D диагностика окончательно расставляет все точки над “i”, есть ли хромосомные аномалии или их нет. Со 100% точностью можно утверждать, имеются ли пороки развития нервной системы, скелетная дисплазия, заячья губа или волчья пасть.
А как складываются отношения Роберта и Кристины с другими вашими детьми?
Марина А.: Старший сын, Леон, ждал братика. Целовал живот, когда я была беременна. Он с такой нежностью к нему относится с самого рождения. Во время кормлений, когда я через трубку заливала Роберту молочко, Леон сидел рядом и держал братика за руку, очень ответственно подходил к этому процессу. А ведь у них разница в возрасте меньше двух лет. И сейчас он так радуется успехам брата. Например, когда Роб пытается стоять, Леон прибегает и рассказывает мне: «Ты представляешь, Роберт стоял! Я считал до трёх, а он стоял!» Я очень рада, что у Роберта есть старший брат, он огромная поддержка для всех.

Леон с младшим братом Робертом
Марина В.: Кристина — мой пятый ребёнок, и другие дети видят, что она отличается, задают вопросы. Я считаю, что главное в отношении с детьми — честность, поэтому я им всё объясняю. Они мне помогают следить за ней, играют с ней, делают вместе развивающие зарядки. Волнуются за неё, когда ей нужно уколы ставить. Мы их называем уколами силы. Вся семья вовлечена.
Синдром Прадера-Вилли причины
Причина возникновения заболевания заключается в процессе делеции и мутации родительской копии гена. Несущей копией является 15-ая хромосома. Как правило, из-за мутаций и других генетических процессов ее частица может быть утеряна. Другие генетические процессы включают: транслокации хромосом и генные делеции, случайные мутации, униотцовскую дисомию.
Импринтинг воздействует на вышеперечисленные гены посредством снижения или удаления активности материнских копий, таким образом, выражаются только родительские копии. Рассматриваемая патология является результатом потери копии родительского гена.
Процесс делеции материнской хромосомы этого же участка является предтечей появления синдрома Ангельмана, которая наряду с болезнью Прадера-Вилли является первой в медицинской науке, описанной патологиями импринтинга человека.
Вероятность проявления болезненных процессов у новорожденного в семье, где уже есть носитель данного заболевания, связан с генетическим механизмом, который дал ход расстройству. Риск появления синдрома у новорожденного составляет менее одного процента, если у ребенка нормально протекают процессы делеции гена и униотцовской дисомии. В случае мутации региона, связанного с импринтингом, вероятность заболевания вырастает до пятидесяти процентов.
Транслокации хромосом в двадцати пяти процентах случаев приводят к проявлению синдрома у следующего ребенка. Диагностика вышеперечисленных процессов и механизмов происходит посредством пренатального тестирования.
Наблюдения, проводимые с участием людей и животных, показывают, что возникновение СПВ напрямую связано с удалением 29 генных копий.
Синдром Ди Джорджи
При синдроме Ди Джорджи у больных отмечается наличие врожденной формы аплазии паращитовидных желез и тимуса. Является разновидностью идиопатического изолированного гипопаратиреоза. Встречается достаточно редко.
При этом заболевании патологические изменения касаются околощитовидных (паращитовидных) желез, у которых отмечается дисгенез или агенезия. Вилочковая железа (тимус) отсутствует от рождения. В результате сочетания таких патологий происходит резкое снижение числа Т-лимфоцитов, формируется иммунологическая недостаточность. Кроме того, этот синдром сопровождается формированием врожденных аномалий крупных сосудов.
Заболевание является аутосомным и определяется наличие мутации в 22 хромосоме. В большинстве случаев причиной является спорадическая делеция 22q11 (реже микроделеция 22q11.2). Наследование происходит по доминантному принципу, с полом не связано. Некоторые авторы не соглашаются с такой характеристикой и приводят аргументы в пользу аутосомно-рецессивного типа, обладающего различной эспрессивностью.
Для заболевания характерно нарушение процесса эмбриогенеза 3-4 жаберных карманов, что приводит к нарушению закладки вилочковой железы и паращитовидных желез.
В клинике наиболее постоянными симптомами являются кандидомикоз и гипопаратиреоз, довольно часто сопровождающиеся нарушением процесса формирования рта, носа и ушей.
Тимус из-за нарушения развития в эмбриональном периоде остается неразвитым. Эпителий тимуса не обеспечивает нормального процесса развития Т-клеток. В итоге формируется специфическая форма иммунодефицита, при которой ослабляется гуморальный иммунный ответ и ответ на клеточном уровне. Если у ребенка имеется подобное патологическое нарушение иммунитета, то он будет обладать повышенной чувствительностью к инфекциям бактериального, вирусного и грибкового происхождения.
Синдром может протекать в форме генетически обусловленного отсутствия паращитовидных желез или изолированной недостаточности околощитовидных желез – в сопровождении гипокальциемических судорог, которые начинаются от рождения. Иммунологическая недостаточность приводит к появлению различных инфекционных заболеваний. Как правило, совокупность симптомов вызывает сердечную недостаточность. Кроме того, летальный исход вызывают инфекционные болезни.
Диагностика синдрома предполагает выявление типичных для синдрома патологий: искажения формы лица и черепа, наличие иммунологической недостаточности, аплазии тимуса, дисгенезии или агенезии паращитовидных желез. Ярче всего при заболевании проявляются кандидомикоз и гипопаратиреоз.
Синдром Прадера-Вилли
Синдром Прадера-Вилли- врожденное заболевание, при котором возникает сочетание ожирения, низкого роста, снижения функции половых желез (гипогонадизм) и низкого интеллекта. Это заболевание имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьировать от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека.
Заболевание впервые описано швейцарскими педиатрами А. Prader и H. Willi в 1956 г. и встречается у 1 человека на 25000-10000 новорожденных. Причиной данного генетического заболевания является отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15 отцовской хромосоме. Следует отметить, что с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно. Для этого применяются специальные цитогенетические и молекулярно-генетические методы.
Дети с синдромом Прадера — Вилли обычно рождаются доношенными с незначительной внутриутробной гипотрофией и нередко в асфиксии. В 10-40% случаев наблюдается ягодичное предлежание. Заболевание характеризуется выраженной мышечной гипотонией при рождении, сохраняющейся в течение первого года жизни ребенка. Сосательный и глотательный рефлексы снижены, что затрудняет кормление ребенка. Из-за гипотонии у таких детей задерживается развитие двигательных функций: они с трудом учатся держать голову, сидеть и т. д. Мышечная гипотония постепенно уменьшается и к школьному возрасту почти полностью исчезает.
Позднее, к второму-четвертому году жизни появляются постоянное чувство голода и отсутствие насыщения, приводящие к развитию ожирения, причем отложение жира наблюдается преимущественно на туловище и в проксимальных отделах конечностей. Из-за тяжелого ожирения грозным осложнением является обструктивное апноэ (остановка дыхания) во сне.
Рост больных нередко снижен. Часто отмечается долихоцефалия (удлиненная форма головы), миндалевидный разрез глаз, низко расположенные ушные раковины, широкая переносица, маленький рот с тонкой верхней губой. Стопы и кисти больных диспропорционально маленькие (акромикрия). У 75% детей наблюдается слабая пигментация кожи, волос и радужки.
У мальчиков при рождении отмечается недоразвитие полового члена, мошонки,крипторхизм, а у девочек недоразвитие половых губ, иногда и матки. В дальнейшем заболевание проявляется задержкой или отсутствием полового созревания, бесплодием.
Психомоторное развитие отстает от возрастной нормы — коэффициент интеллектуального развития — от 20 до 80 ед. (при норме 85-115 ед.). Как правило, дети с синдромом Прадера-Вилли имеют хорошую долговременную зрительную память, они могут научиться читать, могут обладать богатым пассивным словарем, но их собственная речь обычно хуже, чем понимание. Слуховая память, математические навыки и навыки письма, зрительная и слуховая кратковременная память у таких детей обычно значительно хуже. Больные доброжелательны, настроение характеризуется частой сменой. Описаны нарушения координации, судороги, косоглазие. Продолжительность жизни больных может достигать 60 лет и более. Нередко у таких детей развивается сахарный диабет.
Лечение Синдром Прадера-Вилли является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако если диагностировать данное заболевание на раннем этапе и начать его лечение, то прогноз развития заболевания становится более оптимистичным.
Младенцы со сниженным мышечным тонусом должны получать массаж и другие виды специальной терапии. Комплекс лечебных мероприятий включает также диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). Рекомендуется терапия гормоном роста.
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Медико-генетическое консультирование Родителям ребенка с синдромом Прадера-Вилли рекомендуется пройти генетическое обследование, прежде чем планировать дальнейшую беременность, поскольку существует риск того, что следующий ребенок у тех же родителей родится также с синдромом Прадера-Вилли, что зависит от механизма, вызвавшего генетический сбой.
Другие симптомы
В дальнейшем данная патология характеризуется следующими симптомами:
- Искривление позвоночного столба.
- Кариес молочных зубов и повышение густоты слюны.
- Склонность к перееданиям.
- Гипофункции половых желез, которые в дальнейшем приводят к бесплодию.
- Ожирение.
- Задержка моторики и речевого развития.
- Отсталость в психомоторном развитии.
- Запоздалость в половом созревании.
Данные симптомы определяются визуально. В подростковый период выявляются следующие симптомы:
- Задержка речевых навыков.
- Избыточный вес при очень низком росте.
- Неестественная гибкость тела.
- Снижение интеллекта и неспособность к обучению.
Диагностика
Он традиционно характеризуется гипотонией, низким ростом, гиперфагией, ожирением, поведенческими проблемами (особенно поведением, подобным обсессивно-компульсивному расстройству ), маленькими руками и ногами, гипогонадизмом и легкой умственной отсталостью. Однако при ранней диагностике и раннем лечении (например, с помощью терапии гормоном роста) прогноз для людей с СПВ начинает меняться. Как и аутизм, СПВ представляет собой расстройство спектра, симптомы которого могут варьироваться от легких до тяжелых и могут меняться на протяжении всей жизни человека. Поражаются различные системы органов.
Традиционно СПВ диагностировали на основании клинических проявлений. В настоящее время синдром диагностируется с помощью генетического тестирования; Рекомендуется тестирование новорожденным с выраженной гипотонией. Ранняя диагностика СПВ позволяет своевременно вмешаться и назначить гормон роста на ранней стадии . Детям с СПВ показаны ежедневные инъекции рекомбинантного гормона роста (ГР). GH поддерживает линейный рост и увеличение мышечной массы, а также может уменьшить озабоченность едой и прибавку в весе.
Основой диагностики является генетическое тестирование , в частности, тестирование метилирования на основе ДНК для выявления отсутствия отцовской области PWS / AS на хромосоме 15q11-q13. Такое тестирование выявляет более 97% случаев
Тестирование, специфичное для метилирования, важно для подтверждения диагноза СПВ у всех людей, но особенно у тех, кто слишком молод, чтобы проявлять признаки, достаточные для постановки диагноза на клинических основаниях, или у тех людей, у которых есть атипичные результаты.
PWS часто ошибочно диагностируется как другие синдромы из-за того, что многие в медицинском сообществе не знакомы с ним. Иногда его ошибочно принимают за синдром Дауна просто из-за относительной частоты синдрома Дауна по сравнению с PWS.
Признаки Синдрома Прадера-Вилли
Дисплазия тазобедренных суставов; ожирение; склонность к перееданию (чаще проявляется к 2-м годам); пониженный мышечный тонус (гипотонус); пониженная координация движений; маленькие кисти и стопы, низкий рост; повышенная сонливость; страбизм (косоглазие); сколиоз (искривление позвоночника); пониженная плотность костей;густая слюна; плохие зубы; сниженная функция половых желёз (гипогонадизм); в результате, как правило, бесплодие; речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики.
Более позднее половое созревание.