«мы были будто бы во сне»: как живут дети со сма после приёма самого дорогого лекарства в мире
Содержание:
Симптомы у ребёнка и взрослого
Основным признаком болезни СМА является мышечная вялость, слабость и атрофия. Однако у каждой из форм спинальных амиотрофий существуют свои особенности:
- При заболевании Вердинга — Гоффмана первые симптомы могут быть обнаружены ещё во время беременности на УЗИ осмотре, так как плод очень слабо шевелится. После родов отмечается невозможность ребёнка самостоятельно держать голову, переворачиваться и позднее сидеть. Почти всё время малыш лежит в расслабленной позе на спине, не имея возможности свести ноги и руки. Также отмечаются частые проблемы с кормлением, так как младенец испытывает трудности с глотанием. Дыхание зачастую нарушено из-за атрофии рёберной мускулатуры. Практически 70% детей погибают, не дожив до двух лет. После диагностики выявляется недостаточная сформированность передних рогов спинного мозга. Если пациент доживает до 7–10 лет, то у него нарастает выраженность мышечной атрофии и он погибает от острой сердечной, лёгочной недостаточности или из-за проблем с пищеварением. В редких случаях больные доживают до 30 лет, и то исключительно при более позднем начале проявления симптомов (около 2 лет).
- При втором типе спинальной мышечной атрофии ребёнок зачастую может самостоятельно дышать и глотать пищу. Однако со временем происходит прогрессирование процесса, и в более старшем возрасте дети оказываются прикованными к инвалидным креслам. Обычно родители начинают замечать, что ребёнок часто спотыкается, падает и у него подгибаются колени. Постепенная невозможность самостоятельно проглатывать пищу появляется с возрастом. Также по мере взросления начинает проявляться сильно выраженное искривление позвоночника (сколиоз). Эта форма считается относительно доброкачественной и позволяет пациентам прожить до старости. В некоторых случаях женщины даже могут выносить и родить ребёнка, однако велик шанс передачи болезни по наследству. При правильном уходе и благодаря регулярным занятиям лечебной физкультурой пациенты могут очень долгое время сохранять дееспособность.
- Ювенильная амиотрофия Кюгельберга — Веландера может впервые регистрироваться в возрасте от двух до восемнадцати лет. На самом раннем этапе симптомы могут отсутствовать, ребёнок полноценно развивается. Постепенно начинает появляться слабость в проксимальных отделах тела, чаще всего в плечах и предплечье. В течение многих лет пациент способен самостоятельно передвигаться и обслуживать себя. Часто наблюдаются мышечные подёргивания (фасцикуляции). Основной пик проявления симптомов регистрируется в возрасте от двух до пяти лет, когда ребёнку вдруг становится сложно бегать, вставать с кровати и подниматься по лестнице. Течение болезни относительно доброкачественное, так как пациент может длительно сохранять возможность самостоятельно передвигаться.
- Бульбоспинальная мышечная атрофия Кеннеди — заболевание, сцепленное с полом, передаётся с Х хромосомой и проявляется исключительно у мужчин во взрослом возрасте. Прогрессирует болезнь медленно и начинается со слабости в мышцах бёдер, затем через 10–15 лет постепенно присоединяются бульбарные расстройства (поражения черепных нервов: языкоглоточного, блуждающего и подъязычного). Так как течение заболевания крайне медленное, то важные функции практически не успевают нарушаться и продолжительность жизни сильно не сокращается. Очень часто болезнь сопровождается эндокринными патологиями: атрофией яичек, снижением либидо, сахарным диабетом.
- Дистальная СМА Дюшена — Арана обычно регистрируется в возрасте 18–20 лет. Первыми поражаются кисти рук, затем полностью верхние конечности. В течение длительного времени постепенно наступает атрофия мышц ног. В крайне редких случаях заболевание останавливается на парезе одной из рук.
- Скапуло-перонеальная спинально-мышечная атрофия Вюльпиана впервые дает о себе знать в старшем возрасте (20–40 лет). Проявляется постепенной атрофией мышц плечевого пояса и разгибателей стопы и голени. Прогноз относительно благоприятный, так как, даже спустя 30 лет с момента начала заболевания, у пациента сохраняется возможность передвигаться самостоятельно.
СМА у беременных связана со множеством осложнений. Зачастую самостоятельно родить женщина не может и ей назначают кесарево сечение.
На рентгеновских снимках видно искривление позвоночника и последующее его исправление с помощью операции
Клинические проявления
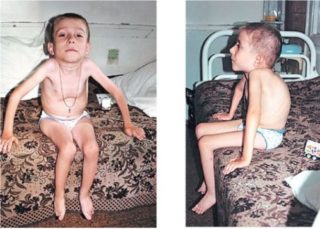
Клинические проявления весьма разнообразны по тяжести и течению. Наряду с тяжелыми формами болезни, протекающими с развитием мышечной слабости и расстройствами дыхания вскоре после рождения, с нарушениями, приводящими к смерти в первые годы жизни, имеются относительно доброкачественные формы, при которых больные доживают до юношеского или зрелого возраста.
Различают раннюю детскую (врожденную), а также детскую и поздние формы заболевания.
Ранняя детская форма амиотрофии клинически может проявляться во внутриутробном периоде или на первом году жизни. Отмечается позднее и вялое шевеление плода. Ребенок рождается с двигательными нарушениями, почти полным отсутствием движений в первые месяцы жизни. При обследовании обнаруживается отсутствие или снижение сухожильных рефлексов, атрофия мышц, разболтанность суставов. Врожденная миатония (см.) описана Оппенгеймом (Н. Oppenheim, 1900). По мнению автора, считавшего ее самостоятельной болезнью, в отличие от амиотрофии Верднига—Гоффманна, при этой форме не отмечается прогрессирования. Ведущим симптомом является атония мышц, мышечная слабость в проксимальных отделах конечностей и туловища, сухожильные рефлексы снижены. Однако многие современные авторы полагают, что врожденная миатония является доброкачественным вариантом ранней формы амиотрофии Верднига—Гоффманна. При ранней детской форме продолжительность жизни от 1 до 7 лет.
Детская форма амиотрофии начинается в возрасте до 4 лет, отличается от первой более медленным течением. Болезнь носит прогрессирующий характер. Длительность заболевания различна. Летальный исход наступает обычно до 14 лет.
Поздние формы спинальной амиотрофии: ювенильная форма амиотрофии и форма с более поздним началом — амиотрофия Кугельберга—Веландера. Описаны Кугельбергом и Веландером (Е. Kugelberg, L. Welander, 1954).
Заболевание характеризуется медленно прогрессирующей мышечной слабостью, атрофией мышц, наличием фасцикуляций, отсутствием пирамидных симптомов. Передается по аутосомно-рецессивному типу. Встречается у мужчин в 2 раза чаще, чем у женщин. У большинства больных наблюдается слабость проксимальных отделов верхних и нижних конечностей. Атрофия мышц, возникающая во всех случаях, может быть завуалирована наличием жировой клетчатки, часто обнаруживается гипертрофия ягодичных и икроножных мышц. Заболевание медленно прогрессирует, больные живут в среднем до 40 лет, иногда дольше. В более поздних стадиях в патологический процесс вовлекается мускулатура дистальных отделов конечностей, однако течение заболевания доброкачественное и в поздних его стадиях двигательные функции относительно сохранены, больные способны передвигаться, обслуживать себя.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс

Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Клиническая картина
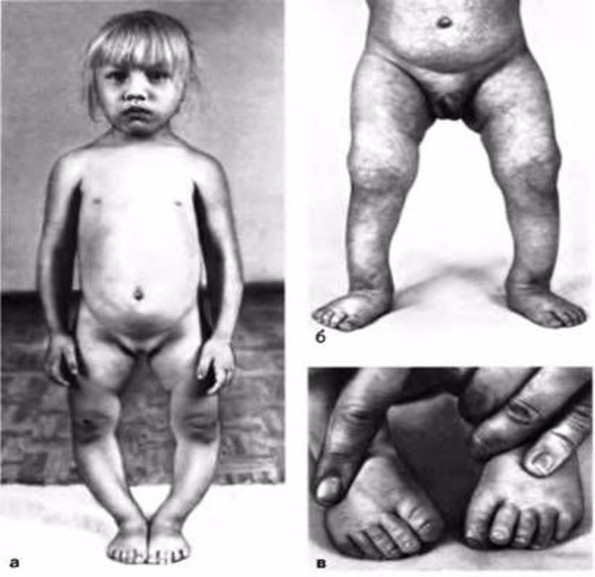
 Ребенок со спинальной мышечной атрофией
Ребенок со спинальной мышечной атрофией
Клинические проявления патологии зависят от возраста, в котором развилось заболевание, и степени тяжести амиотрофии.
Симптомы спинальной мышечной атрофии:
- отсутствие рефлексов в ногах и руках;
- задержка в развитии: позднее сидение, стояние, ходьба или вовсе невозможность осуществления подобных действий;
- занятие позы «лягушка» маленькими детьми: в сидячем положении бедра отведены в стороны, а колени согнуты;
- снижение тонуса дыхательных мышц, слабый кашель, ослабленный крик и плач у новорожденного;
- застойный процесс в органах дыхательной системы, задержка бронхиального секрета, которая вызывает дыхательную недостаточность;
- тремор языка;
- нарушение процесса сосания, глотания, кормления.
 Выделены 4 формы заболевания, каждая из которых имеет свои симптомы:
Выделены 4 формы заболевания, каждая из которых имеет свои симптомы:
- Болезнь Вердинга-Гоффмана (1 тип). Начинает развиваться уже во время пренатального пребывания, первые симптомы возникают в 5-6 месяцев после рождения. Характерные признаки – слабость мышц, снижение рефлексов, выраженное затруднение процесса сосания, дыхания, глотания. Продолжительность жизни – до 1 года в 95% случаев. Летальный исход обусловлен дыхательной недостаточностью.
- Промежуточная амиотрофия Дубовица (2 тип). При заболевании второго типа первые клинические признаки обнаруживают в 3-15 месяцев от рождения. Менее 25% грудничков могут сидеть, но нарушен процесс ходьбы и ползания. Для патологии характерны симптомы паралича, выпадение глубоких сухожильных рефлексов, нарушение глотания. Многие больные уже к 2-3 годам становятся инвалидами и вынуждены находиться в инвалидной коляске. Продолжительность жизни при спинальной мышечной атрофии 2 типа – до 5 лет.
- Спинальная амиотрофия Кугельберг-Веландера (3 тип). Первые симптомы возникают в возрасте 15 месяцев-19 лет и схожи с клинической картиной, присущей 1 типу заболевания. Отличие – медленная прогрессия и лучший прогноз.
- Спинальная амиотрофия 4 типа или взрослая форма. Слабость мышц прогрессирует медленно, в патологический процесс в основном вовлекаются проксимальные мышцы.
У взрослых спинальную амиотрофию разделяют на следующие виды:
- болезнь Кеннеди (амиотрофический боковой склероз);
- болезнь Арана-Дюшенна (хронический полиомиелит взрослых);
- болезнь Шарко-Мари-Тута (моторно-сенсорная нейропатия с поражением дистальных отделов конечностей).
Екатерина Мартынова, мама Артёма
— Мы молодая семья из Воронежа, в мае мне будет 25 лет, а супругу 26. У нас было много планов, другие приоритеты. Но всё поменялось, когда Артёму, нашему единственному ребёнку, исполнилось 11 месяцев и мы узнали о его диагнозе. Речь идёт о начале 2019 года, тогда в мире была зарегистрирована только «Спинраза».
Мы записались в программу раннего доступа к «Спинразе» и получили таким образом шесть уколов. После первых четырёх инъекций Артём перестал слабеть, начали возвращаться навыки переворота на живот, на бок, стал громче кричать, начал сидеть. Курс закончили в мае 2020 года. Если бы мы не получили «Спинразу», то было бы очень страшно.
В мае 2019 в мире зарегистрировали «Золгенсму». Мы, конечно, осознавали, что такую сумму тем более невозможно собрать. Но потом стали встречать истории, когда это всё же удавалось: на Диму Тишунина, на Матвея Чепуштанова. И тоже решили попробовать.
Деньги собирали на протяжении полугода. До начала пандемии COVID-19 успели провести флешмоб, ярмарку на Масленицу, концерт в поддержку сына. Об Артёме говорили на всех городских праздниках, после этого сбор на сутки ускорялся.
Последние три месяца сбора мы сидели дома и работали через соцсети и звонки. Мы волновались, что всё затихнет на карантине, но на самом деле нам пандемия помогла: люди больше времени проводили в интернете и там узнавали об Артёме. Сбор мы закрыли своими силами, без перевода от какого-то одного человека, но нам помогали крупные организации. Например, один из банков перевёл около 12 млн.
Укол «Золгенсмы» Артём получил 30 июня 2020 года, в два года и два дня. Это не наша вина: дистрибьютора задержали с поставкой. Но врач в НИКИ педиатрии им. Вельтищева пошёл нам навстречу, всё-таки два дня — не критичная разница, тут важнее ориентироваться на вес ребёнка. Препарат ввели за час, первое время поднялась температура, но, как объяснили врачи, это обычное явление. Через две недели всё прошло, а через месяц анализы пришли в норму.
Сейчас Артёму два года и восемь месяцев. Он чувствует себя хорошо, крепнет, становится сильнее. Он, конечно, не побежал, не встал, но ползает с поднятой головой, отталкивается ручками и ножками, поднимает корпус. Это для кого-то мелочь, но для нас — прогресс семимильными шагами. «Золгенсма» работает, и это чудо. В Воронеже ходим в бассейн и на ЛФК, скоро полетим в Тюмень делать ортезы.
Наш сын — настоящий герой. Требовательный мальчик, целеустремлённый, шустрый, энергичный, хотя нам это пока не на руку, потому что концентрация на нуле, а нам нужно терпение. Он добрый, сильный, нежный, ласковый, харизматичный. Когда вырастет, будет непростым человеком, как мне кажется.
Болезнь Артёма сильно изменила нас с мужем. Поменялись приоритеты, мы начали понимать, что в этой жизни ценно. Эта ситуация сплотила нашу семью, мы стали больше друг друга ценить, между нами появилась особенная связь. Я сейчас получаю высшее образование, в этом году у меня диплом. На работу не вернусь, но ищу занятие, чтобы можно было совмещать с семьёй. Папа наш предприниматель, амбициозный, теперь у него ещё больше стимула работать. И, конечно, мы помогаем и продолжим помогать другим деткам. Необходимость тратить свои силы и энергию на это я особенно ясно поняла за время сбора. Чем больше отдаём, тем больше приходит
Важно делать это бескорыстно и не ждать чего-то взамен
Диагностика и лечение болезни Верднига-Хофмана
На ранних стадиях заболевания бывает трудно дифференциировать заболевание, так как симптоматика может быть схожа с другими болезнями:
- острый полиомиелит отличается отсутствием прогрессирования заболевания и несимметричными параличами;
- миопатия – также имеет наследственное происхождение, имеет прогрессивное течение, но причиной слабости мышц является нарушение в них обменных процессов;
- врожденная миатония наиболее схожа с болезнью Верднига-Хоффмана, отличить их достаточно можно с помощью биопсии мышечной ткани.
Для диагностики заболевания неврологу понадобятся данные о первом проявлении симптомов, характере их развития, наличия сопутствующих заболеваний.
Проводится ряд исследований для постановки диагноза:
- Электронейромиография выявляет нарушения в работе нервно-мышечной системы. Наблюдаются изменения по мышечному типу, что указывает на патологию двигательного нейтрона;
- Генетический анализ выявляет мутацию гена SMN;
- Биохимия крови на уровень креатинкиназы, показатели в пределах нормы не исключают заболевание;
- Биопсия мышц для морфологического исследования, которое выявляет пучковую атрофию мышечных волокон, чередующихся со здоровыми, а также разрастание соединительной ткани;
- МРТ для исключения других заболеваний.
Для проведения диагностики плода внутриутробно применяется метод биопсии хориона, кордоцентез, амниоцентез. Выявление заболевания является показанием к прерыванию беременности. Вылечить пациента с болезнью Верднига-Хоффмана невозможно. Для продления жизни и улучшения ее качества применяют симптоматическое лечение. Развитие болезни и ухудшение симптомов сдерживают путем обеспечения работы обменных процессов в мышечной ткани.
С помощью лечебной физкультуры и массажа улучшается кровообращение, снижается риск застоев, поддерживается работоспособность мышц, предотвращается неподвижность суставов и потеря ими эластичности. Нагрузки должны быть непродолжительными и осторожными. Физиотерапия способствует удержанию двигательных навыков на имеющемся уровне, их укрепление. Специальные приспособления помогут самостоятельно передвигаться, пользоваться компьютером и даже писать. Портативные аппараты ИВЛ дают возможность пациентам находиться за пределами стационара и проживать жизнь продуктивнее.
Как диагностировать болезнь?
Появление характерных симптомов может указывать на развитие СМА, однако для подтверждения диагноза необходимо генетическое исследование. В 95% случаев заболевание связано с мутацией, представляющей собой полную или частичную делецию гена SMN1. Как правило, чтобы установить диагноз, достаточно выявить данную делецию4.
Для генетического тестирования обычно необходим образец крови.
Иногда проводится анализ крови на содержание креатинфоскиназы (КФК) —фермента, высвобождающегося из разрушающихся мышц. Однако этот тест неспецифичен, поскольку повышение уровня КФК свойственно не только СМА, но и многим другим нервно-мышечным заболеваниям, например, болезни Помпе.
В некоторых случаях может назначаться:
- Биопсия мышечного волокна;
- Электромиография, помогающая оценить уровень биоэлектрических потенциалов, которые возникают в мышцах.
Носителям делеции SMN1, которые могут передать мутацию по наследству, может быть рекомендована предимплантационная генетическая диагностика, используемая для скрининга пораженных СМА эмбрионов (при экстракорпоральном оплодотворении), а также пренатальное тестирование. Последнее включает анализ ворсин хориона, бесклеточный анализ ДНК плода и другие методики.
Суды и сборы
По данным руководителя фонда «Семьи СМА» Ольги Германенко, по всему миру лечение «Спинразой» получили 10 тыс. человек. В России по состоянию на середину декабря начали или продолжили инъекции этого препарата 209 человек, из них восемь взрослых.
«Показатель хороший, но недостаточный», — констатирует она.
При этом, по словам Ольги Германенко, именно в 2020 году некоторые семьи пациентов со СМА столкнулись с затягиванием процесса назначения препарата, в том числе и через суд. Среди доводов региональных властей — неопределённость источников и порядка финансирования закупок, отсутствие «Спинразы» в перечне жизненно необходимых и важных лекарственных препаратов (ЖНВЛП), а также малая эффективность терапии.
24 ноября 2020 года «Спинраза» была внесена в перечень ЖНВЛП. Теперь государство может контролировать стоимость лекарств в перечне, а региональные минздравы лишились возможности отказывать в предоставлении дорогостоящего препарата под предлогом его отсутствия в списке.
Кроме того, производитель «Спинразы» выразил готовность снизить её цену на 25%, таким образом, каждый четвёртый пациент сможет лечиться за счёт этой экономии. Также на основании перечня ЖНВЛП формируются так называемые территориальные программы медицинской помощи — значит, регионам будет проще закладывать соответствующий бюджет.
Через два дня после включения «Спинразы» в перечень жизненно необходимых лекарств произошло ещё одно важное для всех пациентов со СМА событие: в России был зарегистрирован препарат «Эврисди». К тому же появление второго зарегистрированного препарата в перспективе может ещё снизить цены на рынке
К тому же появление второго зарегистрированного препарата в перспективе может ещё снизить цены на рынке.
Регистрация в России самого дорогого лекарства от СМА, «Золгенсмы», ожидается в 2021 году. Пока этого не произошло, семьи пациентов либо собирают деньги на лечение в частном порядке (единичные сборы даже удалось закрыть), либо судятся за право получить терапию за счёт государства.
Особенность «Золгенсмы» в том, что, согласно инструкции, препарат надо применить до того, как ребёнку исполнится два года, тогда как терапию «Спинразой» и «Эврисди» можно начать в любом возрасте.
Опираясь именно на этот довод, суд отказал в предоставлении «Золгенсмы» Косте Гепалову из Санкт-Петербурга, которому в сентябре исполнилось два года. Однако летом специалисты в ЕС, а с ними и американские медики пришли к выводу, что важнее критерий веса ребёнка, а не его возраста. Семья Гепаловых подала повторный иск в суд с новыми документами и ожидает решения.
Биохимические исследования
При амиотрофии установлено умеренное повышение активности ферментов: альдолазы, креатинфосфокиназы, трансаминаз в сыворотке крови. Однако по сравнению с первичными миопатиями (см.) это повышение выражено значительно слабее. Другим характерным биохимическим признаком амиотрофии является креатинурия, отражающая степень атрофии мышечной ткани; одновременно у больных снижается экскреция с мочой креатинина. Креатининовый показатель мочи, представляющий отношение креатинина к сумме креатина и креатинина и в норме равный единице, снижается до 0,8 (С. Н. Давиденков). Содержание в плазме крови и в моче у больных амиотрофией свободных аминокислот существенно не меняется. Наследственные амиотрофии следует дифференцировать с мышечными атрофиями (см. Атрофия мышечная). Прогноз неблагоприятный при ранних и быстротекущих формах амиотрофии — больные погибают в детском возрасте. При поздних и медленно текущих формах прогноз относительно благоприятный: больные благодаря сохранным мышечным группам могут компенсировать функцию пораженных мышц. Следует избегать переохлаждения и переутомления, ведущих к обострению заболевания.
Родителям, имеющим ребенка, больного спинальной или невральной амиотрофией, рекомендуется воздержание от дальнейшего деторождения.
Симптомы разных форм болезни
Существует общий набор признаков СМА, по которым можно заподозрить патологию, если других проблем не обнаруживается, либо диагноз вызывает сомнение. Группу симптомов сводят к проявлению вялого периферического паралича:
- выраженная мышечная слабость или атрофия разных мышечных групп;
- сначала в процесс вовлекаются конечности – симметрично, ноги, а затем руки, постепенно втягивается и туловища;
- отсутствуют расстройства чувствительности и тазовые нарушения;
- наиболее выраженные проблемы затрагивают проксимальные или дистальные мышечные группы.
У пациентов появляются подергивания и фибрилляции – мерцательные аритмии.
Признаки СМА1
Заболевание Верднига-Гоффмана бывает 3 видов:
- Врожденная форма. Начинается в течение 1-6 месяца жизни, обладает тяжелыми симптомами. Обнаружить признаки можно во внутриутробном развитии – эмбрион будет мало двигаться. Гипотония наблюдается сразу же после рождения ребенка. Такие младенцы не держат голову, не могут сидеть. Постоянно находятся в позе лягушки с раздвинутыми конечностями. Сначала симптомы появляются в ногах, затем в руках, после этого страдает дыхательная мускулатура. Психическое развитие у таких детей медленное, они редко доживают до 2 лет.
- Ранняя спинальная мышечная атрофия. Первые признаки начинают беспокоить пациента до 1,5 лет, чаще всего – после какой-либо инфекции. Даже если раньше ребенок мог стоять и сидеть, теперь он утрачивает эти функции. Развиваются парезы, а затем поражаются дыхательные мышцы. Ребенок умирает, как правило, в результате затяжной пневмонии или дыхательной недостаточности в возрасте 3-5 лет.
- Поздняя форма. Патология встречается после 1,5 лет, двигательные возможности сохраняются у ребенка до 10 лет. Медленный прогресс симптомов приводит к дыхательной недостаточности и смерти в возрасте до 18 лет.
Признаки болезни Кугельберга-Веландера
Возникает в возрасте от 2 до 15 лет. Сначала в процесс вовлекаются нижние конечности, затем пояс таза, на последних стадиях страдает плечевой пояс и дыхательная система. Примерно у 25% пациентов появляется синдром псевдогипертрофии мышц, из-за чего патологию путают с мышечной болезнью Беккера.
Спинальная мышечная атрофия Кугельберга-Веландера не сопровождается костными деформациями, и пациенты способны сами себя обслуживать в течение долгих лет.
Амиотрофия Кеннеди
Эта патология входит во взрослую группу, болеют представители мужского пола после 30 лет. Женщины от патологии не страдают. Течение – умеренное, сначала поражаются ножные мышцы, последующие 10-20 лет пациент сохраняет привычный ритм жизни. Только после этого начинают страдать мускулы рук и головы. У многих пациентов со временем наступают эндокринные изменения: атрофия яичек, отсутствие либидо, сахарный диабет.
Дистальная СМА
Эта форма спинально-мышечной атрофии также развивается у взрослых пациентов после 20 лет. Ее второе название – СМА Дюшенна-Арана. Риск развития патологии сохраняется вплоть до 50 лет. Атрофия начинается в руках, вызывает синдром «когтистой лапы», потом переходит на крупные мышцы. Со временем появляются парезы мышц нижних конечностей, а туловище страдает редко. Прогноз у этой формы благоприятный, если не присоединяется торсионная дистония или болезнь Паркинсона.
СМА Вюльпиана
Скапуло-перонеальная форма спинально-мышечной атрофии, сопровождаемая симптомом «крылатых» лопаток. Появляется в среднем в 20-40 лет, позже встречается реже. Поражается плечевой пояс, а через некоторое время – руки и нижние конечности. При этой форме спинальной болезни двигательные функции у пациента сохраняются на 30-40 лет.